

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Chemotherapy-induced peripheral neuropathy (CIPN) is one of the adverse effects of chemotherapeutic agents used in cancer treatment, which is often a major reason for stopping or changing anticancer therapy. The incidence of survivors suffering from these neuropathic pain conditions is increasing, due to improvements in the success rates of these agents in cancer treatment and the consequent increases in survival rates [1]. The prevalence of CIPN was reported to be 68.1% in the first month of treatment [2], and can sometimes cause persistent limitation in quality of life, even in cases in which the chemotherapy cured the patient’s cancer [3]. Although treatments for CIPN include anticonvulsants, such as gabapentin, carbamazepine, or lamotrigine, and antidepressants, such as tricyclic antidepressants or serotonin norepinephrine reuptake inhibitors [3], these medications provide only partial relief in many cases, likely because the pathophysiology is not clearly understood.
Among the proposed pathomechanisms of CIPN, proinflammatory processes induced by chemotherapy agents have attracted increasing attention; this not only destroys the tumor cells [4] but may also lead to neuroinflammation, contributing to the development of CIPN. Animal studies have reported increased levels of cytokines, including interleukin (IL)-6, IL-8, IL-1β, and tumor necrosis factor (TNF)-α, which can sensitize nociceptors [5-7]. A rat model of CIPN exhibited infiltration of macrophages in the dorsal root ganglion and activation of microglia in the spinal dorsal horn [8]. Another study showed that pretreatment with the microglia/macrophage inhibitor minocycline prevented paclitaxel-evoked allodynia [9]. Taken together, modulation of the inflammatory response to chemotherapeutic agents may be a useful target for the treatment of CIPN.
Prostaglandin D2 (PGD2) is the most abundant prostaglandin in the brain of rats [10] and humans [11], and plays important roles in the central nervous system (CNS), including roles in regulation of the sleep cycle and body temperature, hormone release, and nociception [12]. Synthesis of PGD2 from the product of cyclooxygenase (COX) activity, prostaglandin H2 (PGH2), is catalyzed by two forms of PGD synthases, hematopoietic (H-PGDS) and lipocalin-type (L-PGDS) [13]. PGD2 is released from cells as a mediator to act upon a specific membrane receptor (DP1 or DP2) in the neighboring neurons and glia [14]; this release was reported to increase in response to systemic and spinal cord inflammatory processes [15], which is a well-established mechanism of CIPN. Furthermore, intrathecal administration of PGD2 receptor antagonist significantly attenuated mechanical allodynia in a rat model of spared nerve injury (SNI) [16]. However, the roles of PGD2 signaling in the pathogenesis, and as a potential therapeutic target in CIPN, have not been investigated.
The present study was performed to examine the role of PGD2 signaling and the receptor subtype in mechanical allodynia using a cisplatin-induced neuropathic pain model in rats. In addition, we investigated the expression patterns of DP1 and DP2, and PGD synthases in the spinal cord.
Go to : 
MATERIALS AND METHODS
1. Animals
The experimental protocol was reviewed and approved by The Institutional Animal Care and Use Committee of Chonnam National University (IACUC No. CNU IACUC-H-2018-48). Adult male Sprague-Dawley rats weighing 160-190 g were housed in a room maintained at a constant temperature (22-23°C) with an alternating 12-hour light/dark cycle and free access to food and water. An intrathecal catheter was implanted for drug administration for each animal under sevoflurane anesthesia, as described previously [17]. Seven days were allowed after the surgery for recovery.
2. Drugs
The following drugs were used in the experiments: cisplatin (Tocris Cookson Ltd., Bristol, UK), AMG853 (5-chloro-4-[2-[(2-chloro-4-cyclopropylphenyl)sulfonyl]amino]-4-[(1,1-dimethylethyl)amino]carbonyl]phenoxy]-2-fluorobenzeneacetic acid; Tocris Cookson Ltd.), MK0524 (5-chloro-4-[2-[(2-chloro-4-cyclopropylphenyl)sulfonyl]amino]-4-[(1,1-dimethylethyl)amino]carbonyl]phenoxy]-2-fluorobenzeneacetic acid; Cayman Chemical, Ann Arbor, MI), and CAY10471 ((+)-3-[(4-fluorophenyl)sulfonyl]methylamino]-1,2,3,4-tetrahydro-9H-carbazole-9-acetic acid; Cayman Chemical). Cisplatin was diluted in 0.4% dimethyl sulfoxide (DMSO) in physiological saline immediately prior to intraperitoneal injection at a final concentration of 0.2 mg/mL. The other drugs were dissolved in 50% DMSO. Experimental drugs were administered intrathecally in a volume of 10 μL through the implanted catheter using a manual gear-operated syringe pump, followed by an additional 10 μL of the vehicle solution for each drug to flush the catheter.
3. Behavioral testing
CIPN was induced by intraperitoneal administration of cisplatin (2 mg/kg/day) once daily for 4 consecutive days as described previously [18]. The von Frey test was performed to measure the mechanical withdrawal threshold, at which a positive response was assumed when brisk withdrawal or flinching of the paw was observed during, or immediately after, applying one of the filaments with logarithmically increasing stiffness (0.4, 0.7, 1.2, 2.0, 3.6, 5.5, 8.5, and 15.0 g). Then, the 50% probability paw withdrawal threshold (PWT) was calculated using the up and down method [19]. The cutoff value was 15 g. Only rats showing mechanical allodynia (50% withdrawal threshold < 4 g) after cisplatin injection were included in this study.
4. Experiment protocol
On the day of the experiment (12 days after CIPN modeling), animals were acclimatized in a box with a wire mesh floor for at least 20 minutes and randomly allocated into the experimental groups for intrathecal delivery of experimental drug or vehicle solution. Behavioral testing was carried out by an investigator blinded to the treatments. To investigate whether PGD2 signaling contributes to mechanical allodynia, the DP1 and DP2 antagonist, AMG853 (100 or 300 μg, n = 8), was administered. Mechanical withdrawal thresholds were measured at 15, 30, 60, 90, 120, 150, and 180 minutes after intrathecal administration. To elucidate the role of the PGD2 receptor subtype in PGD2 signaling, the selective DP1 antagonist, MK0524 (100 or 300 μg, n = 7), or DP2 antagonist, CAY10471 (3, 10, or 30 μg, n = 7), was injected 15 minutes prior to the von Frey test. The doses of the administered drugs were chosen based on pilot experiments.
5. Western blotting
Dorsal halves of rat lumbar 4-6 spinal cord sections were harvested under anesthesia with sevoflurane, and frozen in liquid nitrogen. The samples were homogenized in radioimmunoprecipitation assay lysis buffer, combined with a phosphatase inhibitor cocktail, and centrifuged at 13,000 rpm for 10 minutes at 4°C. The supernatants were collected, and protein concentration was measured with a BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL). Aliquots of 30 μg of protein from each sample were subjected to 10%-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred onto polyvinylidene difluoride membranes at 100 V for 90 minutes. The membranes were blocked with Tris-buffered saline-Tween 20 (TBST) containing 5% non-fat milk for 1 hour at room temperature and incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-DP1 (1:200; Cayman Chemical), rabbit polyclonal anti-DP2 (1:200; Cayman Chemical), rabbit polyclonal anti-prostaglandin D synthase (hematopoietic) (1:800; Abcam, Cambridge, MA), rabbit monoclonal anti-prostaglandin D synthase (lipocalin) (1:1,000; Abcam), and mouse monoclonal anti-β-actin (1:2,000; Cell Signaling Technology, Danvers, MA). Blots were washed with TBST and incubated with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) (1:3,000; Cell Signaling Technology) or horseradish peroxidase-conjugated horse anti-mouse IgG (1:3,000; Cell Signaling Technology) at room temperature for 1 hour. The proteins were detected by enhanced chemiluminescence reagents (Millipore, Bedford, MA) and a computerized image analysis system (Uvitec, Cambridge, UK) was used to quantify the intensity of protein bands on the blots.
6. Statistical analysis
All data are shown as means ± standard error of means. The time-response data are exhibited as the withdrawal threshold in grams. The dose-response data are quantified as the area under the time course curves (AUC) for each dose using the trapezoidal rule, which were analyzed by one-way analysis of variance (ANOVA) with Bonferroni or Dunnett’s T3 post hoc test. The changes in protein expression level in the spinal cord were compared by unpaired t-tests. In all analyses, P < 0.05 was taken to indicate statistical significance.
Go to :
RESULTS
The mechanical PWT decreased significantly after intraperitoneal injection of cisplatin for 4 consecutive days. This characteristic mechanical allodynia persisted for at least 21 days, as reported previously [18]. The baseline threshold before experimental drug administration did not differ among the groups. Intrathecal administration of AMG853 significantly increased the PWT. One-way ANOVA exhibited a statistically significant difference in the AUCs among the three groups (F [2,21] = 7.082, P = 0.004). Post hoc comparisons by the Dunnett’s T3 method showed that the AUCs of the AMG853 100 and 300 μg groups were significantly greater than those of vehicle-treated controls (P = 0.020 and 0.030, respectively, Fig. 1). While intrathecal delivery of MK0524 did not affect pain behavior (Fig. 2), intrathecal CAY10471 significantly increased the PWTs with an effect lasting up to 180 minutes (Fig. 3). One-way ANOVA revealed significant differences among groups in the AUCs of withdrawal threshold (F [3,24] = 23.993, P < 0.001). Post hoc test using Bonferroni correction revealed that the AUCs of CAY10471-treated rats were significantly increased compared to the vehicle-treated controls in a dose-dependent manner (Fig. 3).
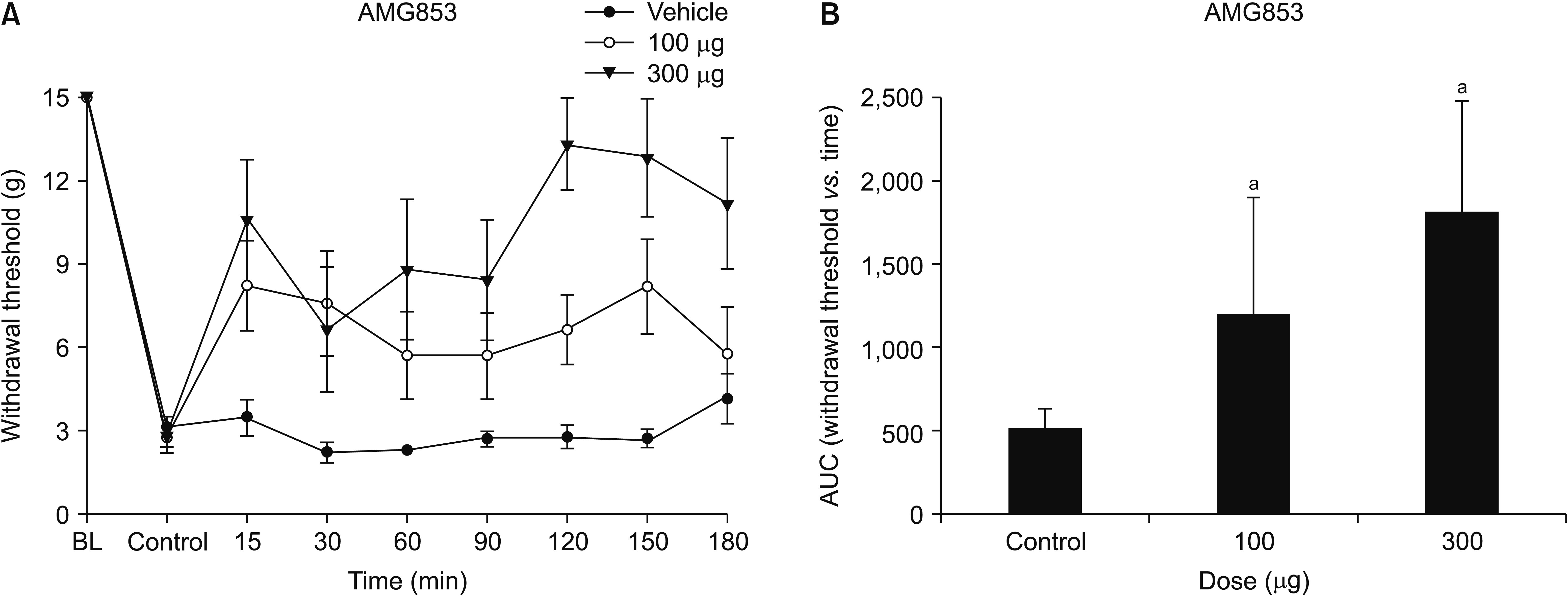 | Fig. 1(A) Time-response and (B) dose-response data showing the effects of the DP1 and DP2 antagonist, AMG853, on the hind paw withdrawal response in cisplatin-treated rats. Data are presented as the mechanical withdrawal thresholds in grams or the area under the time course curve (AUC) for the withdrawal threshold. Each line or bar represents the mean ± standard error of mean of 8 rats. BL: baseline value. aP < 0.05 compared to the vehicle group.
|
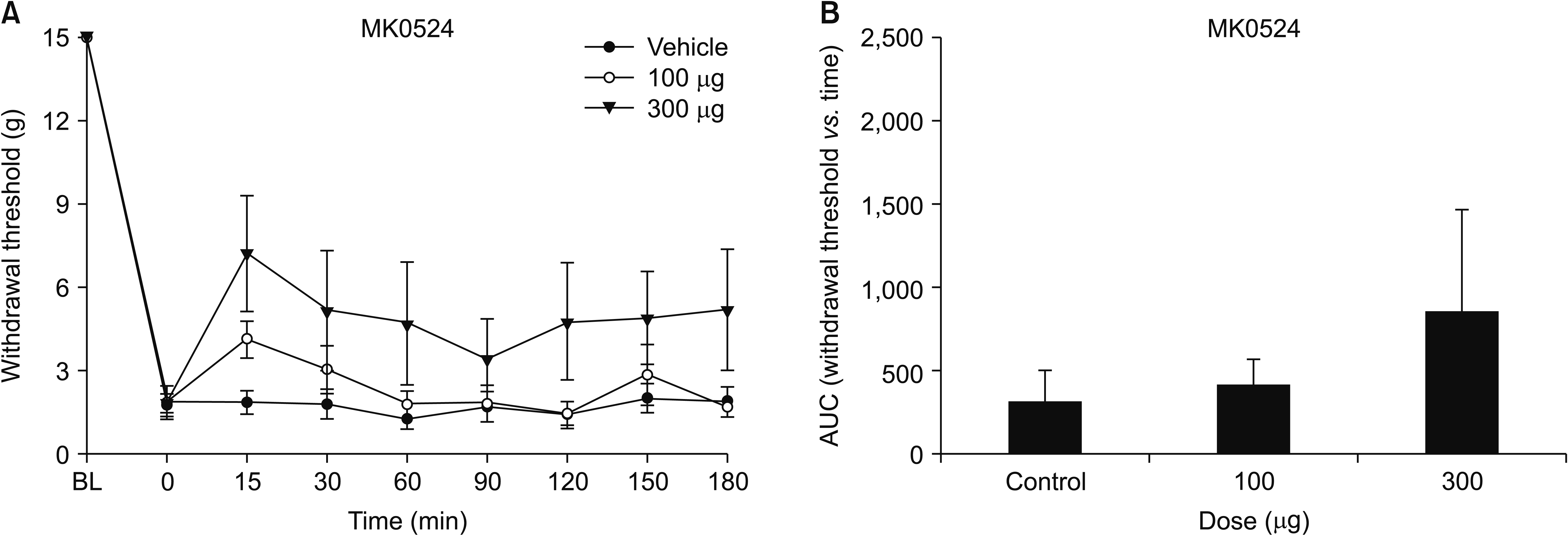 | Fig. 2(A) Time-response and (B) dose-response data showing the effects of the DP1 antagonist, MK0524, on the hind paw withdrawal response in cisplatin-treated rats. Data are presented as the mechanical withdrawal thresholds in grams or the area under the time course curve (AUC) for the withdrawal threshold. Each line or bar represents the mean ± standard error of mean of 7 rats. BL: baseline value.
|
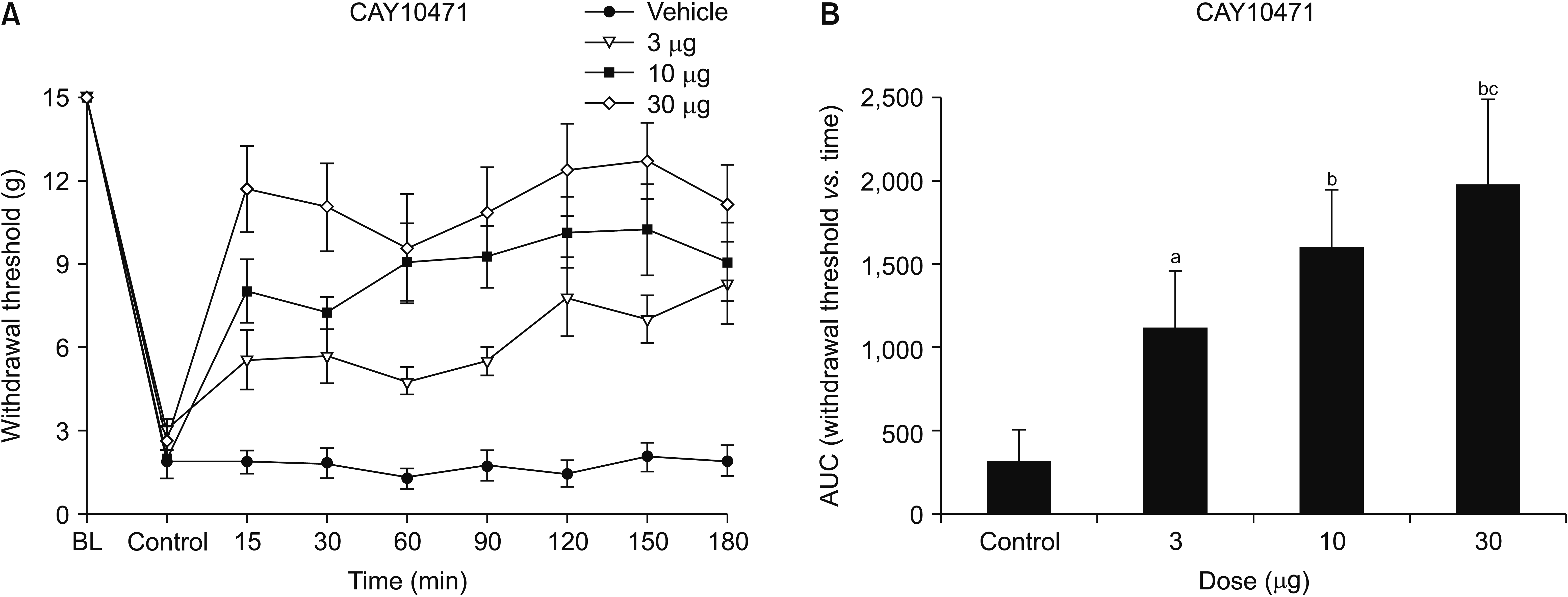 | Fig. 3(A) Time-response and (B) dose-response data showing the effects of the DP2 antagonist, CAY10471, on the hind paw withdrawal response in cisplatin-treated rats. Data are presented as the mechanical withdrawal thresholds in grams or the area under the time course curves (AUC) for the withdrawal threshold. Each line or bar represents the mean ± standard error of mean of 7 rats. BL: baseline value. aP = 0.004 compared to the vehicle group. bP < 0.001 compared to the vehicle group. cP = 0.001 compared to the 3 µg dose group.
|
Western blotting analysis showed comparable expression levels of DP1 and DP2 protein between the spinal cord samples harvested from the cisplatin-treated animals and vehicle-treated controls (Fig. 4A, B). In the CIPN group, the level of L-PGDS protein expression, but not that of H-PGDS, was significantly increased compared to the control group (P < 0.001, Fig. 4C, D).
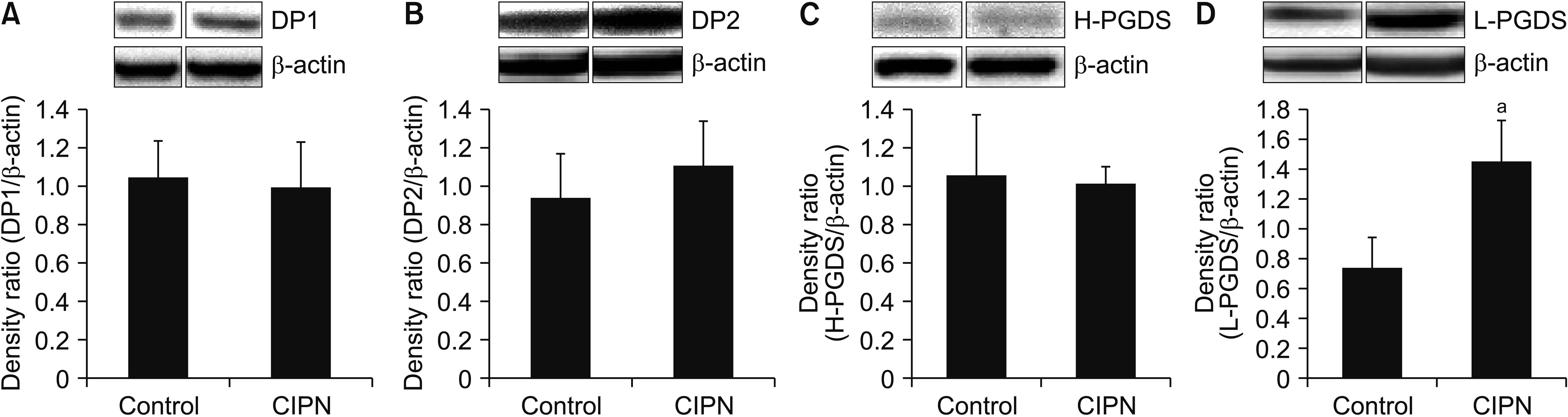 | Fig. 4Expression of (A) DP1 and (B) DP2, (C) hematopoietic prostaglandin synthase (H-PGDS), and (D) lipocalin prostaglandin synthase (L-PGDS) protein by western blotting analyses (optical density [OD]) in the spinal cord of vehicle-treated (control) or cisplatin-treated (chemotherapy-induced peripheral neuropathy [CIPN]) animals. Insets show representative western blots. Lanes 1 and 2 show vehicle- and cisplatin-treated animals, respectively. Histograms show quantification of the OD expressed as a ratio to the corresponding β-actin level. Values are means ± standard error of mean of 6 rats. aP < 0.001 compared to the vehicle group.
|
Go to :
DISCUSSION
This study was performed to evaluate the role of spinal cord PGD2 signaling in CIPN. Blockade of DP2, but not DP1, relieved mechanical allodynia, and expression of the receptor protein was confirmed in the spinal cord of the animals. This speculation was further supported by the increased expression level of L-PGDS protein, which serves as the main mediator in catalyzing the conversion of PGH2 to PGD2 in the CNS [20]. These findings indicate that augmented PGD2 signaling in the spinal cord contributes to mechanical allodynia via DP2 in the cisplatin-induced neuropathic pain model in rats, and that modification of this pathway may present a novel therapeutic target in CIPN.
PGD2 has been shown to act in the peripheral tissue in relation to vasodilatation, bronchoconstriction, inhibition of platelet aggregation, and allergic reactions [21]. Many PGD2 receptor antagonists are currently under clinical investigation for the treatment of allergic diseases and asthma [22]. A recent phase 2 clinical trial of the PGD2 receptor antagonist, fevipiprant, showed promising results in asthma patients [23,24]. In the CNS, PGD2 is the most abundant prostaglandin released from neuronal and non-neuronal cells, including glia and invading immune cells [13]. PGD2 participates in the induction of non-rapid eye movement sleep and plays a regulatory role in the control of body temperature, hormone release, and pain processing [12]. Two types of PGD2 receptors, DP1 and DP2, are known, both of which are expressed in the spinal cord [16]. DP1 stimulates adenylyl cyclase and leads to an increase in cyclic adenosine monophosphate (cAMP) concentration, while DP2 decreases cAMP and increases intracellular calcium [25]. The findings of the present study were consistent with previous reports in different animal models of pain. Minami et al. [26] reported that intrathecal injection of PGD2 provoked hyperalgesia and allodynia. In the rat SNI model, mechanical allodynia was attenuated by intrathecal DP2 antagonist but not DP1 antagonist [16]. Taken together, these observations indicate that PGD2 has a pronociceptive role in pain processing via DP2 in the spinal cord.
Biosynthesis of PGD2 is catalyzed by two synthases, H-PGDS and L-PGDS. H-PGDS is characterized as the spleen-type PGDS acting in a glutathione (GSH)-dependent manner for the synthesis of PGD2 in the peripheral tissues, while L-PGDS is known as the lipocalin-type or GSH-independent PGDS and is expressed primarily in the CNS, heart, kidney, testis, and lung [12]. L-PGDS is one of the major proteins in cerebrospinal fluid [13], and radioimmunoassay and western blotting analyses confirmed the presence of L-PGDS and PGD2 in the lumbar spinal cord of rats [27,28]. A systemic endotoxin-induced inflammatory response in the spinal cord, which was verified by increased spinal TNF-α and IL-1β mRNA, enhanced the expression of L-PGDS protein in the spinal cord [15]. Furthermore, the same study demonstrated increased release of PGD2 in an in vitro superfusion chamber study using the spinal cord from the endotoxin-treated animals. These findings suggested that enhanced biosynthesis of PGD2 can be induced by upregulation of L-PGDS in the spinal cord by systemic inflammation. Notably, the activation of the immune system and the development of neuroinflammatory processes in the spinal cord are hallmarks of CIPN independent of the mechanism of action of chemotherapeutic agents [3,29].
Previous investigations examining the dorsal horn of CIPN animals reported glial hyperactivation, increased microglial expression, and increased proinflammatory cytokines, such as TNF-α and IL-1β, in the spinal cord [7,30]. Glial cells strongly affect neuronal signaling by releasing proinflammatory cytokines, which may, in turn, lead to enhanced excitability of neurons [31]. Therefore, cisplatin-produced proinflammatory processes in the present study may have induced the upregulation of spinal L-PGDS protein expression, which can increase PGD2 biosynthesis. In addition to participating in PGD2 synthesis, L-PGDS functions as a binding protein, possibly acting as an intercellular carrier protein to bring PGD2 to the receptor, which may also increase its elimination half-life [32].
The present study had several limitations. First, the role of the upstream pathway of PGD2 synthesis in CIPN remains to be established. Selective or nonselective inhibition of COX by celecoxib, aspirin, or ibuprofen, which blocks the formation of a substrate of L-PGDS, failed to show antinociceptive effects once allodynia had been established [33]. However, when given at the time of administration of a chemotherapeutic agent with the potential for CIPN, indomethacin and celecoxib suppressed hyperalgesia [34]. Spinal expression of COX-2 and prostaglandin E2 synthase increased rapidly with a peak level 4–6 hours after application of endotoxin, while that of L-PGDS was delayed beginning 24 hours after systemic endotoxin-induced inflammation and continued for 48 hours [32]. These observations suggest that each component participating in the biosynthesis of prostaglandins may play a different role during the time course of the development and maintenance of CIPN. Second, there was no significant change in the expression levels of DP1 and DP2 protein in CIPN animals compared to the control group. Therefore, investigation of alteration in the DP2 receptor function will be necessary to further elucidate the down-stream pathway of PGD2 signaling. Third, we did not investigate the anatomical location of the L-PGDS and PGD2 receptor expression in the spinal cord. Although this issue was not elucidated in this study, it has been suggested that the expression of L-PGDS is increased prominently in non-neuronal cells and that of PGD2 receptors are confined to neurons of the spinal cord, including lamina I and II, following exposure to systemic inflammation [32].
In conclusion, the results presented here indicated that intrathecally administered DP2 antagonist significantly attenuated cisplatin-induced mechanical allodynia, indicating that PGD2 signaling contributes to mechanical allodynia in CIPN via DP2 in the spinal cord. The demonstration of spinal expression of DP1 and DP2 and the upregulation of spinal L-PGDS which raises the possibility of enhanced biosynthesis of PGD2 in the spinal cord further supported the role of PGD2 in CIPN. We suggest that a blockade of DP2 receptor activation may serve as a potential therapeutic target for managing CIPN.
Go to :
 XML Download
XML Download