

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Among the various diseases that accompany chronic pain, complex regional pain syndrome (CRPS), previously called reflex sympathetic dystrophy, is one of the most frustrating for patients and physicians. Various treatment modalities, including medications, physiotherapy, regional anesthesia, and neuromodulation have been tried; however, they have demonstrated limited efficacy, and their use remains controversial [1].
Although many studies have attempted to identify the exact pathogenetic mechanism of CRPS, it remains unclear. A multitude of previous studies on CRPS have primarily focused on the autonomic nervous system, peripheral nervous system, or spinal cord to elucidate the pathophysiology of CRPS. Relatively few studies have investigated the supra-spinal region or the brain in relation to CRPS. Some studies have provided supporting evidence that the brain plays a significant role in the pathophysiology of CRPS. It was reported that patients with CRPS have difficulties in recognizing the exact area or position of the affected body part because they have a distorted perception of their body image [2]. Moreover, patients with CRPS feel pain simply by imagining the movement of the affected limb without actually performing any real movement [3], and even looking at a reflection of the affected body part in the mirror produces pain [4]. The results of functional imaging studies of the brain also provide evidence supporting the role of the brain in the pathophysiology of CRPS [5,6].
Considering that the pain in patients with CRPS persists despite treatment of the peripheral limbs and that the chronic pain is strongly associated with emotions, it is necessary to target the brain to identify the central pathogenetic mechanism of CRPS.
Therefore, some researchers argue that CRPS may be a problem of the central nervous system or a systemic disease involving the neuronal system, rather than a peripheral disease [7-9]. Mirror therapy and graded motor imagery, which aim to train the brain against pain from various diseases, are increasingly being used in the treatment of CRPS with significant clinical improvement [10].
Calcium plays a crucial role in a variety of physiologic processes, including signal transduction and cell growth and proliferation [11]. The calcium-mediated signal is the most important intracellular signal for long-term memory and synaptic plasticity [12]. Calmodulin (CaM), an extensively studied calcium-binding protein, participates in regulating many calcium-mediated physiological processes [13]. As an intermediate messenger protein for various physiologic processes, CaM delivers Ca2+ signals to numerous target proteins and modifies their actions. CaM regulates calcium-calmodulin protein kinase 2 (CaMK2), which is one of the most prominent and abundantly expressed protein kinases in the brain. Once calcium ions enter the cell and stimulate the N-methyl-D-aspartate (NMDA) receptor, CaMK2 is activated through auto-phosphorylation, which in turn phosphorylates various enzymes [14]. The resultant conductivity of the assorted ion channels causes excitation of nerve cells, causing pathogenic pain, such as hyperalgesia and allodynia, as observed in CRPS [15].
Central sensitization is significant in CRPS; hyperalgesia and allodynia, the hallmark of central sensitization, are frequently observed in patients with CRPS [16]. CaMK2, the essential mediator of learning and memory [17], contributes to peripheral [18] and central neuropathic pain [19]. There is evidence suggesting that the expression and activity of CaMK2 increases in various chronic pain models. Additionally, a previous study reported that CaMK2 plays a crucial part in synaptic plasticity [12] and central sensitization in nociceptive pain in the spinal cord [20].
Considering these facts, we can infer that the brain, the central control system for cognitive and emotional functions, is associated with the pain and central sensitization observed in CRPS. In this regard, we postulated that cerebral physiologic processes mediated by CaM/CaMK2 increase in CRPS. Thus, this study aimed to investigate whether peripheral injury can increase calcium-mediated cerebral processes in CRPS.
Go to : 
MATERIALS AND METHODS
1. Experimental animals
The Institutional Animal Care and Use Committee of the Seoul National University Bundang Hospital approved this study (IACUC No. BA 1206-106-042-03). We kept the animals in a facility approved by American Association for Assessment and Accreditation of Laboratory Animal Care International as per the guidelines in the Guide for the Care and Use of Laboratory Animals of the National Research Council. We used specific-pathogen-free male Sprague Dawley rats (Orient Bio Inc., Seongnam, Korea) that were 8 weeks old and weighed 200-250 g as the experimental animals. Each animal was housed in an individual cage and acclimated to the environment for one week before production of the CRPS animal models. The chronic post-ischemia perfusion (CPIP) rat model [21,22], which is a commonly employed animal model for CRPS research, was used in this experiment. For CPIP model production, a tight-fitting Nitrile 70 Durometer O-ring® (O-ring West Inc., Lynnwood, WA) was applied to the left hindpaw, just proximal to the medial malleolus of the isoflurane-anesthetized rats, and left for three hours. It was then removed for reperfusion at the distal part of the O-ring® application site. A sham operation was performed on the control group under similar anesthesia by cutting one side of the O-ring® to avoid ischemia at the ankle.
2. Measuring the withdrawal threshold
The withdrawal threshold (WT) was measured as described in a previous study [23]. Each animal was placed on a wire mesh to assess its WT. After twenty minutes of acclimation, we applied mechanical stimulation on the plantar surface of the hindpaw using von Frey hairs (Stoelting Co., Wood Dale, IL) via apertures created in the mesh floor of the cage.
Rapid flinching movements or abrupt withdrawals of the paw after the mechanical stimulation were considered as positive responses, after which we applied the next lighter von Frey hair. In case there was a lack of response upon von Frey hair application, the next heavier hair was applied. Based on a previous study [24], the WT was measured, and the change in WT (ΔWT, %) was calculated using the equation below:
The WT was measured at 1 hour, 4 hours, 1 day, 2 days, 7 days, and 21 days after the CPIP procedure.
On day 21 after the CPIP procedure, the rats were classified into three groups based on the ΔWT, as follows:
A CRPS-positive (P) group (rats with a ΔWT ≥ 50%), a CRPS-negative (N) group (rats with a ΔWT < 50%), and a control group. The criterion of a ΔWT of 50% was thought to be appropriate during the 21-day experiment, and this criterion was also used in other studies that used the CPIP animal model [25-27].
3. Western blot analysis for CaM/CaMK2 expression
Western blot analysis for cerebral CaM/CaMK2 was conducted using samples from animals in the control, P, and N groups (n = 4 rats per group). After euthanasia and decapitation under isoflurane anesthesia, we harvested the right half of the cerebrum of each animal. The tissue samples were sonicated in the T-PER™ reagent (Thermo Fisher Scientific Inc., Rockford, IL), and the extracted proteins were quantified using the Pierce™ BCA protein assay system (Thermo Fisher Scientific Inc.). Total protein was separated using 10%-16% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (200 V, 40 min) and the protein contents were electro-transferred (25 V, 100 min) to polyvinylidene difluoride membranes. Following this, the membrane was blocked in Tris-buffered saline/Tween-20 (TBS/T, 0.1%) containing 5% skimmed milk for two hours at room temperature. This was followed by probing the membranes using antibodies overnight at 4℃. The membranes were washed four times for 10 minutes in the TBS/T and incubated for two hours at room temperature with a secondary antibody conjugated with horseradish peroxidase. Protein bands were visualized using an enhanced chemiluminescence kit after washing four times with the TBS/T. The intensities of the protein bands were quantified using ImageJ densitometry software (National Institutes of Health, Bethesda, MD). The primary antibodies used were as follows:
Rabbit anti-calcium/CaM monoclonal antibody (Abcam, Cambridge, UK), rabbit anti-CaMK2 monoclonal antibody (MilliporeSigma, Burlington, MA), and mouse anti-β-actin monoclonal antibody (Sigma-Aldrich Corp., St. Louis, MO). We used peroxidase-labeled anti-rabbit immunoglobulin G and anti-mouse immunoglobulin G (Vector Laboratories Inc., Burlingame, CA) as the secondary antibodies.
4. Statistical analysis
Statistical analysis was performed on the data obtained using SigmaPlot® ver. 12.0 (Systat Software Inc., Chicago, IL). Data are expressed as the mean ± standard error of the mean. Because of the small sample size, we used the Kruskal–Wallis test, followed by Dunnett’s test as the post hoc analysis to compare the ΔWT of the hind paw and CaM/CaMK2 expression levels in the animals for each group at each time point. P values less than 0.05 were considered to reflect statistical significance.
Go to :
RESULTS
The health status of each animal was monitored throughout the experiment by investigators and veterinary staff according to the Guide for the Care and Use of Laboratory Animals of the National Research Council. The animals were free of all viral, bacterial, and parasitic pathogens. A total of 14 animals (7 per group) were selected and allocated to groups P and N after measuring the ΔWT. In addition, seven animals were included in the control group. Finally, 21 animals had their ΔWT values analyzed. Among them, 12 animals (4 per group) were randomly selected for western blot analysis. Before the application of the O-ring®, no differences were found in the WT among the three groups. However, at day 21 after the CPIP procedure, group P showed a significant reduction in the WT compared with groups N and C (group P: 0.89 ± 0.39 g, group N: 12.14 ± 4.18 g, and group C: 14.40 ± 1.94 g; P < 0.01). Fig. 1 illustrates the WT changes before and after the application of the O-ring®.
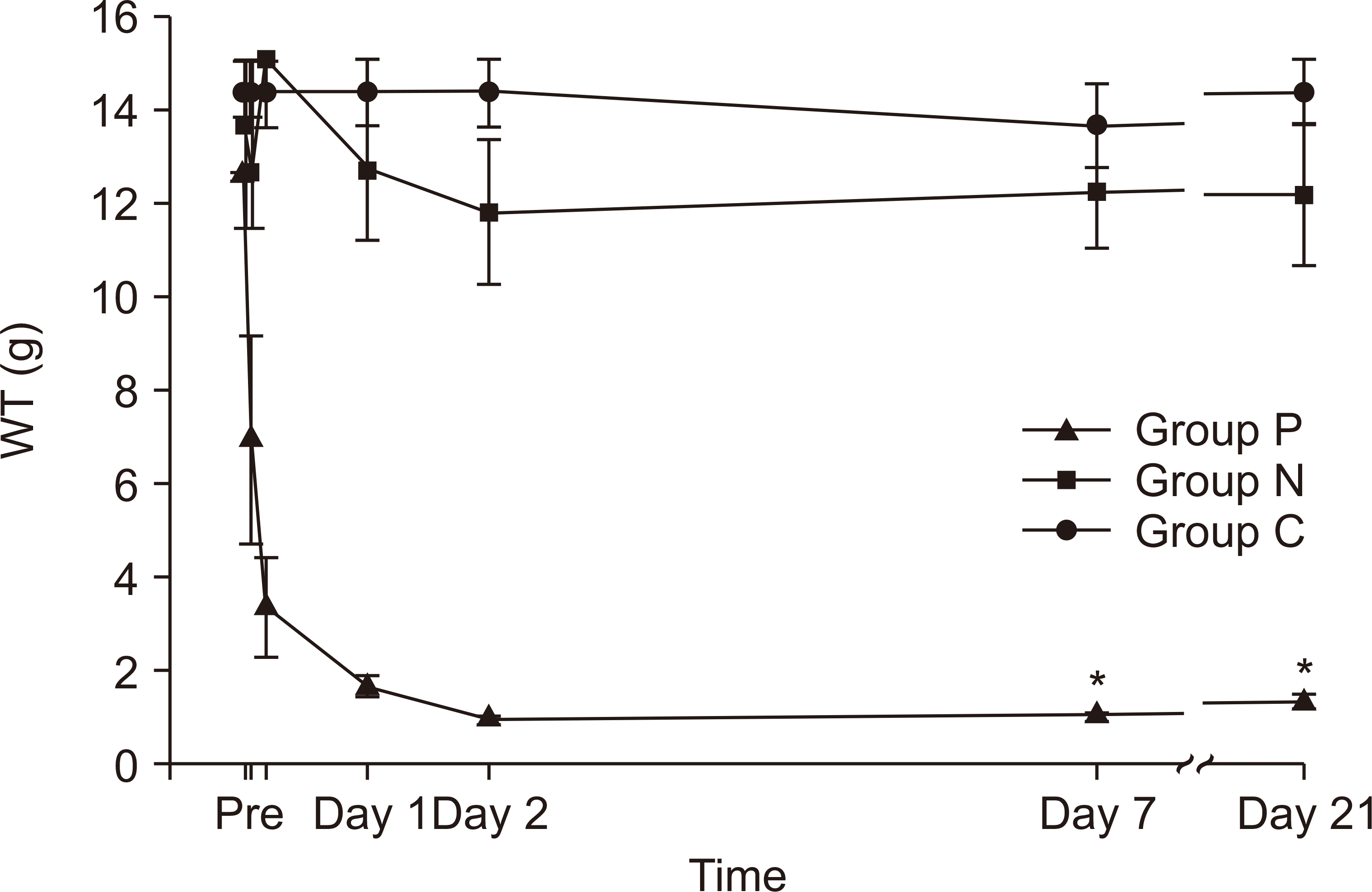 | Fig. 1The WT after CRPS model generation. The WT was measured at 1 hr, 4 hr, 1 day, 2 days, 7 days, and 21 days after the CPIP procedure. Rats in group P showed a significant difference in WT in comparison to those in groups N and C (n = 7 rats per group, P < 0.01). Groups P, N, and C indicate the CRPS-positive (ΔWT ≥ 50%), CRPS-negative (ΔWT < 50%), and control groups, respectively. WT: withdrawal threshold, CRPS: complex regional pain syndrome, CPIP: chronic post-ischemia perfusion. *P < 0.05.
|
Figs. 2, 3 illustrate the results of the western blots for cerebral CaM and CaMK2 expression levels in each group (n = 4 rats per group). The expression of cerebral CaM was significantly elevated in group P compared to groups N and C (group P: 1.05 ± 0.04, group N: 0.93 ± 0.05, and group C: 0.90 ± 0.08; P = 0.013, Fig. 2). Moreover, a significant elevation in the expression of cerebral CaMK2 was observed in group P compared to in groups N and C (group P: 1.22 ± 0.10, group N: 0.96 ± 0.14, and group C: 1.04 ± 0.06; P = 0.021, Fig. 3). However, there was no significant difference in CaM and CaMK2 expression between groups N and C.
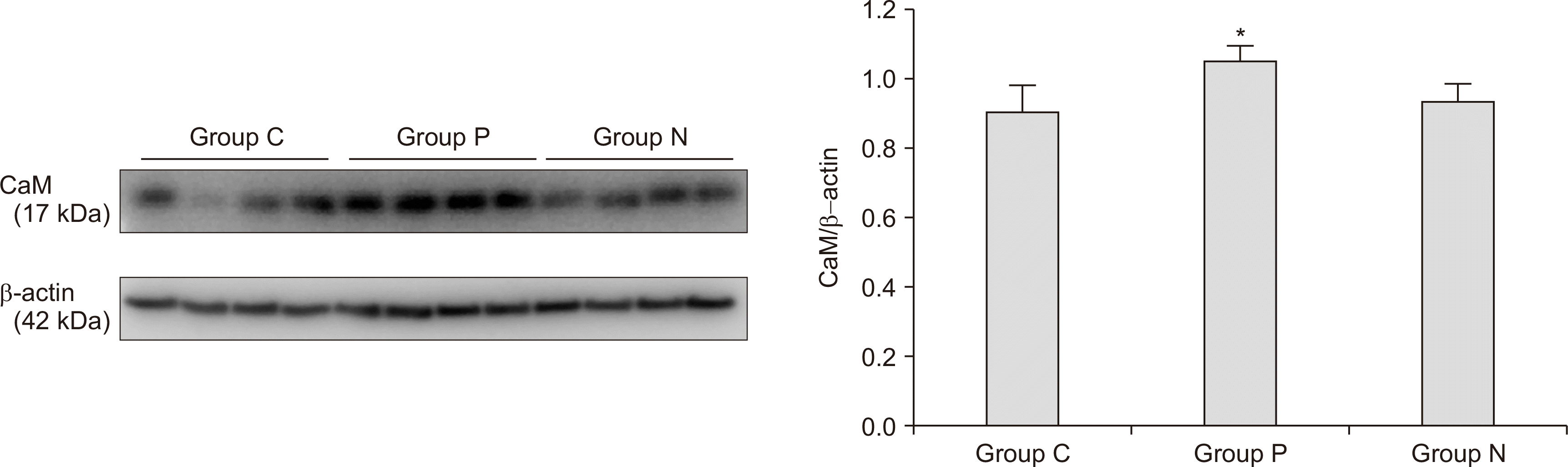 | Fig. 2Expression of cerebral calcium-calmodulin (CaM). A significant increase in the cerebral CaM level was found in group P in comparison to in groups N and C (n = 4 rats per group, P = 0.013). Error bars indicate standard errors. Groups P, N, and C indicate the CRPS-positive (ΔWT ≥ 50%), CRPS-negative (ΔWT < 50%), and control groups, respectively. *P < 0.05.
|
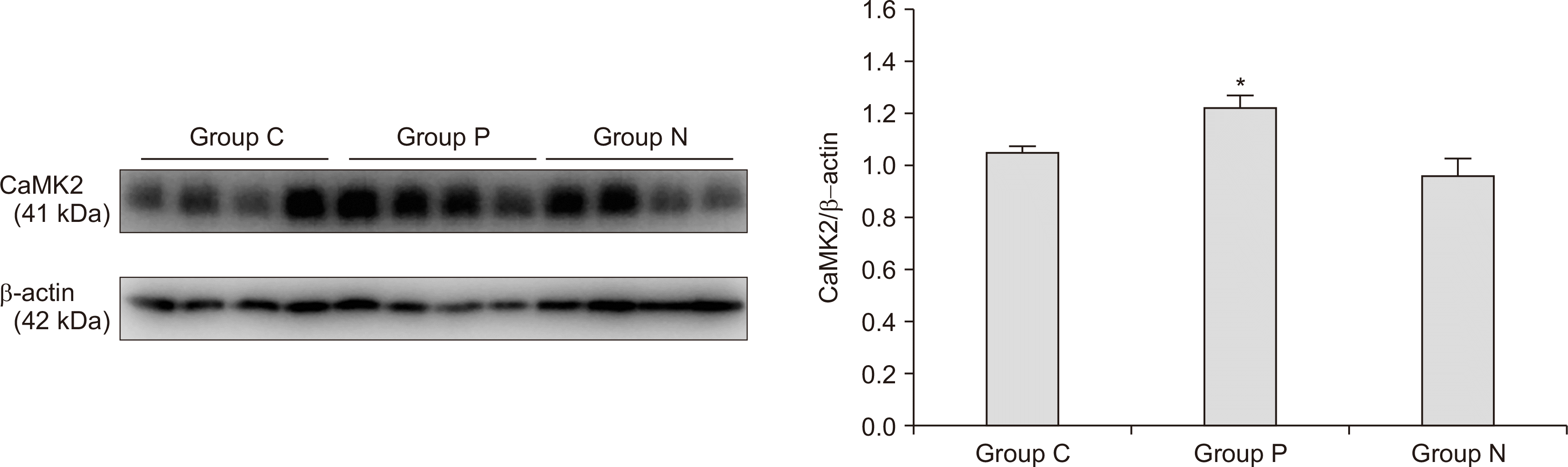 | Fig. 3Expression of cerebral calcium-calmodulin kinase 2 (CaMK2). A significant increase in the cerebral CaMK2 level was found in group P compared to in groups N and C (n = 4 rats per group, P = 0.021). Error bars indicate standard errors. Groups P, N, and C indicate the CRPS-positive (ΔWT ≥ 50%), CRPS-negative (ΔWT < 50%), and control groups, respectively. *P < 0.05.
|
Go to :
DISCUSSION
We found that the expression of cerebral CaM/CaMK2 increased in the CRPS animal models. This finding suggests that peripheral injury can induce calcium-mediated physiological processes in the cerebrum in patients with CRPS. Given that minor peripheral trauma is the primary cause of CRPS, this result indicates the need for further studies on the cerebral changes involved in CRPS.
CaMK2 is considered to be essential in central sensitization [20]. The calcium-CaM complex regulates CaMK2, which is involved in several functions, including neurotransmitter release [28], controlling ion channel activity [29], synaptic plasticity [12], and learning and memory [17]. Moreover, CaMK2 plays roles in central neuropathic pain [18] and long-term potentiation (LTP) [30]. LTP, which shares many aspects with central sensitization [31], is initiated once the NMDA receptors permit Ca2+ into the postsynaptic neuron, and this in turn activates CaMK2. It has been shown that LTP in nociceptive spinal pathways has some characteristics as hyperalgesia [32], with evidence also demonstrating possibly similar mechanisms in pain and memory that contribute to central sensitization and LTP [31]. Based on this information, the finding of cerebral CaMK2 activation might implicate its cerebral involvement in CRPS. There is emerging evidence demonstrating that CaMK2 might also be a new drug target for the treatment of chronic pain, as blocking CaMK2 reduces pain in various painful conditions [33]. Therefore, novel analgesic medications with high specificity for CaMK2 may be developed and used as alternative therapeutic options for intractable pain.
Meanwhile, CaM is an extensively studied Ca2+ binding protein that is expressed in all eukaryotic cells. It plays a crucial role as a multi-functional intermediate messenger protein by transducing Ca2+ signals [34]. CaM also mediates many essential processes such as metabolism [35], neuronal plasticity [36], regulation of cell proliferation [11], and cancer [37]. Many of the calcium-mediated events occur when the released calcium ions bind to and activate CaM. Moreover, CaM antagonists block the capsaicin and NMDA receptors, resulting in pain alleviation [38]. Therefore, the increased cerebral CaM levels observed in our study indicate that calcium-mediated cerebral processes are enhanced in CPIP animal models. Additionally, the increase in both CaM and CaMK2 levels seems to be involved in the pathogenesis of pain in CRPS. It has been suggested that CaMK2, once activated by CaM, can function as a protein switch after peripheral stimulation [39]. The elevation in intracellular Ca2+ through the NMDA receptors after noxious stimuli can trigger the activation of a series of various kinases, including CaMK2. The up-regulation of CaMK2 phosphorylation results in the activation of cyclic adenosine monophosphate response element-binding protein (CREB) [40], thereby regulating the transcription of numerous genes, including the genes for c-Fos and brain-derived neurotrophic factor, which are closely linked to pain and central sensitization [41]. Furthermore, enhancement of the CaMK2-CREB signaling pathway participates in the affective-motivational pain response [42].
This study showed some different result with the previous proteomic identification of cerebral proteins in the CRPS animal model [26]. The previous study showed elevations in inositol 1,4,5-triphosphate receptors, which are regulated by CaM, but no significant increases in CaMK2, as were observed herein. However, in the previous study, CaMK2 was listed as an increased cerebral protein among the 454 proteins that showed expression changes in CRPS, with a P value of 0.086. The lack of a significant finding in that study might have been due to the protein identification method used; the previous study used multidimensional protein identification technology (MudPIT), and the difference in protein expression was searched using a protein database. The MudPIT mainly aims to screen the candidate proteins and does not provide a confirmative result. Therefore, further confirmative analyses, e.g., western blots or enzyme-linked immunosorbent assays, are usually required.
In this study, we used the CPIP model, which is a novel animal model for CRPS that can reproduce the signs and symptoms of CRPS type 1 [21]. Taking into account that the signs and symptoms of CRPS occur without direct nerve injury or fracture after minor tissue damage, this model is an adequate animal model for CRPS research [21].
There are several limitations to this study. First, the cerebral regions that showed increased CaM/CaMK2 expression were not specified in this study. Due to the huge size of the cerebrum and the absence of information about its functional map in the rat cerebrum, we could not specify the areas of the cerebrum involved in CRPS pathogenesis in rats. Functional imaging studies have reported that the activation patterns in patients with CRPS differ from those in healthy individuals in the sensorimotor cortex, secondary somatosensory cortex, and inferior frontal gyrus [43]. Future studies aimed at identifying the specific areas of the cerebrum involved in the pathogenesis of CRPS are needed. Second, the exact roles of the augmented CaM/CaMK2 in the cerebrum are unclear. The effects of CaM/CaMK2 in the development of CRPS require further investigation. Given that increased CaM/CaMK2 is associated with synaptic plasticity and LTP in the central nervous system, we can postulate that increased levels of cerebral CaM/CaMK2 may be associated with the development and maintenance of CRPS. Lastly, we assessed the WT and CaM/CaMK2 expression levels without utilizing antagonists. It would be much more persuasive if pharmacological interventions had been conducted. This is because we only wanted to explore whether calcium-mediated cerebral processes increase after peripheral injury in CRPS. The fact that CaM/CaMK2 increased only in group P and was unchanged in group N suggests that the pain and elevated CaM/CaMK2 may be related. The elevated expression of cerebral CaM/CaMK2 observed in this study might encourage further investigations to discover the role calcium-mediated cerebral mechanisms play in CRPS pathogenesis. Further studies using CaM/CaMK2 antagonists are needed to investigate the exact supraspinal mechanism of CRPS.
In conclusion, the results of this study suggest that calcium-mediated cerebral processes increase after peripheral injury in CRPS, and that CaM/CaMK2 are possibly involved in central sensitization after peripheral trauma. Further studies on the CaM/CaMK2 molecular pathway in CRPS are needed to reveal the mechanisms underlying CRPS and to find new drug targets for its management.
Go to :
 XML Download
XML Download