

 PDF
PDF Citation
Citation Print
Print


Introduction
All-trans retinoic acid (ATRA) is synthesized from retinol (Vitamin A) and serves as the physiological ligand for retinoic acid receptor (RAR). ATRA binds to the RAR and activates transcription of target genes. Many RAR target genes are involved in myeloid cell differentiation (1, 2), including C/EBPɛ, PU.1 and HOX proteins. In Acute promyelocytic leukemia (APL), characterized by the expression of a PML/RARα (promyelocytic leukemia/RAR alpha) fusion protein, RAR signaling is impaired and myeloid differentiation is arrested at the promyelocytic stage (3). Pharmacologic doses of ATRA induce a conformational change in PML/RARα, resulting in the dissociation of co-repressor complexes and the recruitment of co-activator complexes (4, 5). This allows the restoration of gene transcription involved in myeloid differentiation. In contrast to APL, other forms of acute myeloid leukemia (AML) are either non-responsive or show blunted responses to ATRA. The underlying reasons identified thus far are associated with impaired RARα function by aberrantly expressed proteins and epigenomic changes that affect RAR signaling (6). Therefore, complementary treatment strategies have been studied to improve the effectiveness of ATRA (7–9).
The transcription factor c-Myc plays essential roles in cell proliferation, apoptosis, and differentiation through its ability to both positively and negatively regulate gene expression (10). c-Myc is frequently overexpressed in AML mainly due to trisomy 8, FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) mutation, or gene amplification and has been shown to contribute to leukemogenesis (11). A recent study has shown that c-Myc binds RARα and represses ATRA-target genes in AML cells (12).
Although c-Myc plays an important role in leukemogenesis, it has been difficult to target pharmacologically. Recently, bromodomain and extra-terminal (BET) domain protein BRD4 emerged as new therapeutic target to suppress c-Myc expression (13, 14). BRD4 recruits positive transcription elongation factor b (P-TEFb) complex to RNA polymerase II and initiates transcriptional elongation (15). Bet bromodomain inhibitors, such as JQ1 and I-BET151, competitively bind to acetyl-lysine recognition pocket of BRD4, displaces BRD4 from chromatin, and represses transcription of genes in the affected chromatin region (16). In c-Myc-driven malignancies such as multiple myeloma and AML, Bet bromodomain inhibitors repress c-Myc expression and thereby exhibit significant anti-cancer effect (13, 14). Therefore, Bet bromodomain inhibitors have emerged as promising anticancer agents, currently being evaluated in clinical trials in hematologic malignancies with some encouraging results (17, 18). However, treatment with Bet bromodomain inhibitor produced the dose-limiting toxicities and resistance mechanisms were identified (19–21). Thus, increasing efforts are needed to identify rational targeted drug combination for synergy and to overcome potential resistance mechanisms. Several combinations have been explored with BET inhibitors and synergistic effects were observed with greater inhibition of proliferation, compared to either drug alone (22–24).
In this study, we investigated the antileukemic effects of ATRA and BET bromodomain inhibitor JQ1 when they were used in combination against non-APL AML cells. We observed that the combination of JQ1 and ATRA synergistically reduced the proliferation of AML cells. The enhanced growth inhibition might be associated with inhibition of c-Myc and its target gene, hTERT.
Go to : 
Materials and Methods
Cell culture
HL-60 and MV4-11 cell lines were purchased from American Type Culture Collection (ATCC) and cultured according to ATCC recommendations. All cell lines were maintained in an incubator at 37°C and 5% CO2. All experiments with cell lines were performed within 6 months after thawing or obtaining from ATCC.
Reagents
All-trans-retinoic acid (ATRA) was purchased from Sigma (St Louis, MO). JQ1 was purchased from Selleck chemicals (Houston, TX). They were all dissolved by dimethyl sulfoxide (DMSO) as a stock solution at 50 mM and 10 mM respectively.
Cell proliferation
Cells were seeded at a density of 1×105 cells to 2×105 cells per ml and rested for 5 hours. Cells were then treated with ATRA and/or JQ1 for the indicated days. The number of viable cells was assessed using the Cell counting kit-8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. The coefficient of drug interaction (CDI) is calculated as follows: CDI=AB/(A×B). According to the absorbance of each group, AB is the ratio of the combination groups to control group; A or B is the ratio of the single agent group to control group. Thus, CDI value<1, =1 or >1 indicates that the drugs are synergistic, additive or antagonistic, respectively.
Flow cytometry
Myeloid differentiation was assessed by detection of the cell surface CD11b expression. Cells were pre-incubated with 0.5 μg Fc block (BD Biosciences, San Jose, CA) for 10 minutes at RT and stained with anti-CD11b-PE antibody (BD Biosciences, San Jose, CA) for 30 min at 4°C. The labeled cells were then washed and analyzed for CD11b expression. The apoptosis assay was performed using Annexin V-FITC Apoptosis Detection Kit (Abcam, Cambridge, UK). Briefly, cells were washed with staining buffer and resuspended in 300 μl of binding buffer. 100 μl of the samples were incubated with FITC-labeled Annexin V for 15 min in the dark at room temperature. The reaction was stopped by adding 300 μl of binding buffer and analyzed. Flow cytometry was performed on a FACSCalibur equipped with CellQuest Pro software (BD systems, San Jose, CA). FlowJo software (Version7.6.1, Treestar, Ashland, OR) was used for subsequent analysis.
Western blotting
Cells were washed with ice-cold PBS and lysed in RIPA buffer (Thermo Scientific, Waltham, MA) containing Complete protease inhibitor cocktail tablets (Roche, Indianapolis, IN) and PhosSTP phosphatase inhibitor cocktail (Roche, Indianapolis, IN). After incubation on ice for 30 minutes, cell lysates were centrifuged at 13,000 rpm for 15 minutes at 4°C. Supernatants were collected and protein concentrations were determined by Bradford’s protein assay (Bio-Rad, Hercules, CA) according to manufacturer’s protocol. 20 μg protein extracts were subjected to 8%~12% SDS-PAGE gels according to molecular weight of detected protein bands and then transferred onto polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, CA). The blots were blocked for 1 h in 5% bovine serum albumin in Tris-buffered saline containing 0.1% Tween-20 (TBS-T), and incubated with primary antibody at 4°C overnight. The blots were washed three times and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (Cell Signaling Technology, Danvers, MA) at room temperature for 1 h. After washing, blots were developed by enhanced chemiluminescence (ECL) according to manufacturer’s instructions (Cell Signaling Technology, Danvers, MA) and imaged with a LAS-4000 imaging system (Hugifilm Life Science). The following primary antibodies were used: anti-c-Myc (Abcam, Cambridge, UK). anti-C/EBPɛ (GeneTex. Irvine, CA). anti-β-actin (Cell Signaling Technology, Danvers, MA).
RNA extraction and Real-time PCR
Total RNA was extracted from cells with the Trizol reagent (Invitrogen), and cDNA was synthesized using High Capacity RNA-to-cDNA kits (Applied Biosystems, Foster City, CA). Real-time PCR was performed in duplicate using SYBR green reagents (Roche, Indianapolis, IN). The PCR protocol consisted of thermal cycling as follows: pre-amplification at 95°C for 2 min followed by 45 cycles of 95°C for 10 sec; 60°C for 10 sec; and 72°C for 10 sec using LightCycler 96 System (Roche, Indianapolis, IN). The target mRNA expression was quantified using the Δ ΔCT method and normalized to GAPDH expression.
Go to :
Results
JQ1 synergizes with ATRA to reduce proliferation of AML cells
We first explored the ability of ATRA to inhibit the proliferation of HL-60 and MV4-11 cells by CCK-8 assay. Both the HL-60 and MV4-11 cells were exposed to increasing concentrations of ATRA for the indicated days. ATRA concentrations of 20~100 nM induced similar growth inhibition in both cell lines (Fig. 1A). Therefore, we selected a dose of 100 nM ATRA which has been used in most in vitro studies (7–9). To determine the combined effects of ATRA and JQ1 on AML cell proliferation, we selected the dose of 50 nM and 100 nM JQ1 which were not enough for complete growth inhibition.
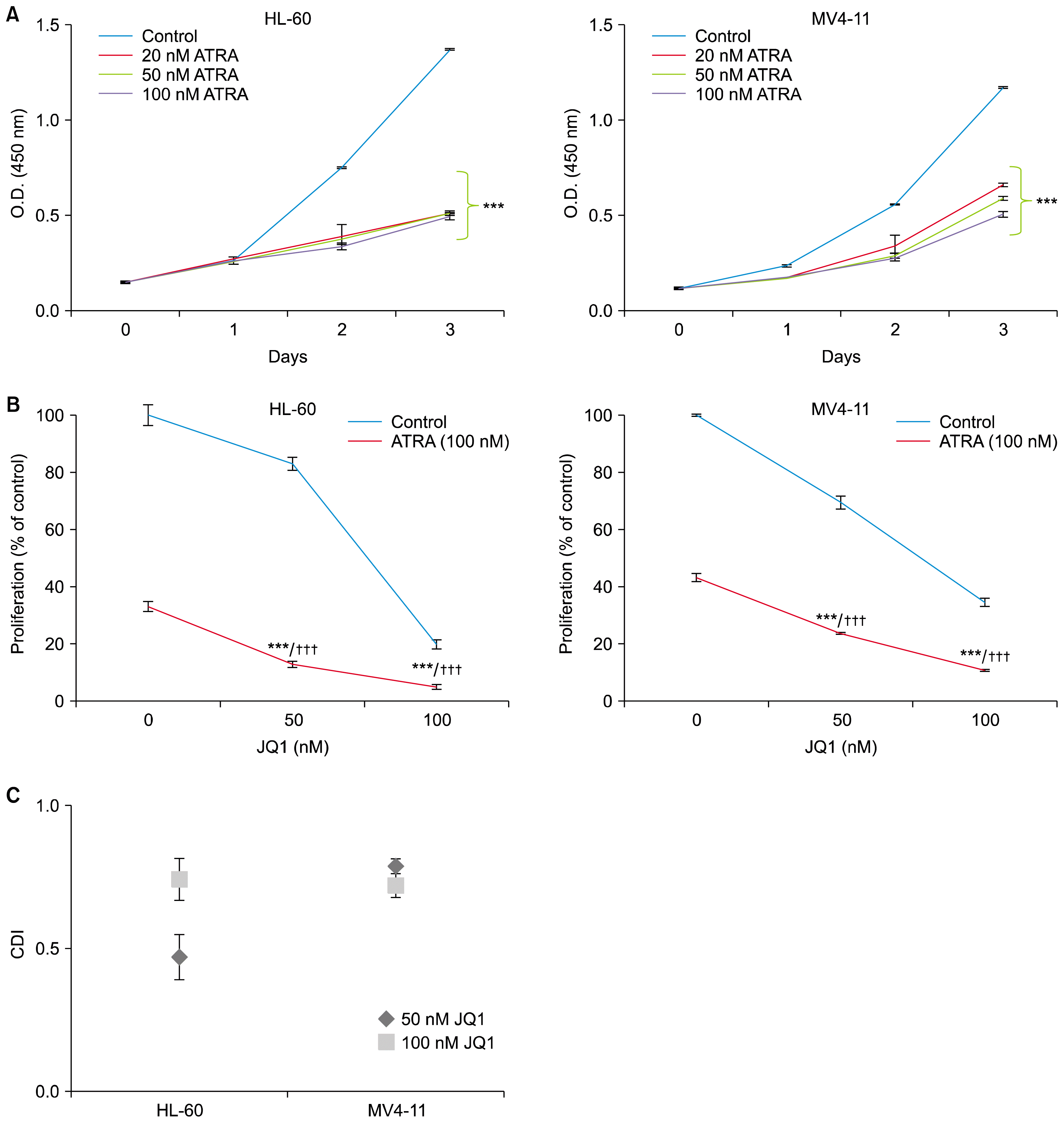 | Fig. 1JQ1 synergizes with ATRA to reduce proliferation of AML cells. (A) HL-60 cells and MV4-11 cells were treated with ATRA (20~100 nM) for the indicated days. Cell proliferation was measured using a CCK-8 assay. The data represent the means±SD of 3 independent experiments. *ATRA versus control (***p<0.001). (B) HL-60 cells and MV4-11 cells were treated with the indicated concentrations of JQ1 and/or 100 nM ATRA for 3 days. Cell proliferation was measured using a CCK-8 assay. The data represent the means±SD of 3 independent experiments. *combination versus ATRA (***p<0.001); †combination versus JQ1 (†††p<0.001). (C) CDI values at different concentrations of JQ1 and 100 nM ATRA were calculated as described in materials and methods.
|
As shown in Fig. 1B, both HL-60 and MV4-11 cells were treated with indicated concentrations of JQ1 alone, ATRA (100 nM) alone, or the combination. To test the combination effect, the Coefficient and Drug Interaction (CDI) was calculated as described in materials and methods. As expected from the CCK-8 assay results (Fig. 1B), CDI values less than 1 indicated the combination of JQ1 and ATRA yielded synergistic interactions at both concentrations (50 nM and 100 nM) of JQ1 (Fig. 1C).
JQ1 enhances ATRA-induced differentiation of HL-60 cells
As the combination of JQ1 and ATRA synergistically inhibited proliferation of HL-60 and MV4-11 cells, we sought to examine whether the synergy resulted from differentiation. We assessed myeloid differentiation by increased CD11b expression using flow cytometry. Treatment of HL-60 cells with 100 nM of ATRA alone for 3 days resulted in 41.6±13.3 MFI of CD11b expression. When cells were treated with 50 nM or 100 nM JQ1 alone, MFI of CD11b expression was close to control or minimal respectively. However, combined treatment of ATRA with JQ1 for 3 days significantly induced CD11b expression dose dependently (96.8±16 MFI for 50 nM JQ1 and 142.3±9.7 MFI for 100 nM JQ1) (Fig. 2A). In MV4-11 cells, neither agent alone nor the combination induced CD11b expression (data not shown).
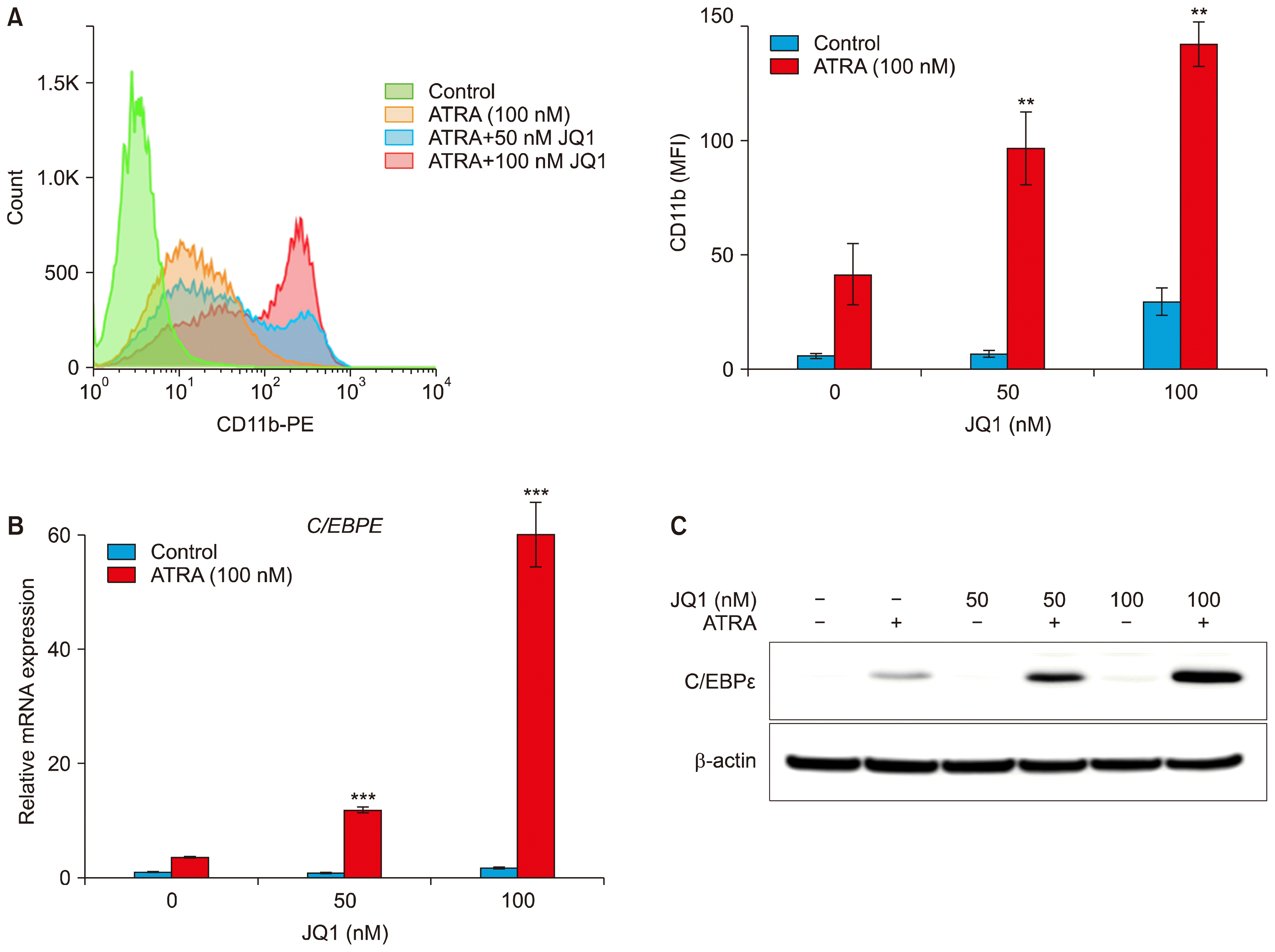 | Fig. 2Effect of JQ1 on ATRA-induced HL-60 cell differentiation. HL-60 cells were treated with the indicated concentrations of JQ1 and/or 100 nM ATRA for 3 days. (A) Histogram (left) and bar graph (right) represented CD11b expression. Values were expressed as Mean fluorescence of intensity (MFI). Experiments were independently repeated three times and results were shown as mean±SD. (B) The mRNA levels of C/EBPɛ were determined by real-time PCR. The relative expression of C/EBPɛ mRNA was normalized to GAPDH and was reported as a percentage of untreated cells. (C) The protein levels of C/EBPɛ were measured by Western blot analysis using 20 μg protein. Similar results were obtained in three independent experiments. *combination versus ATRA (***p<0.001; **p<0.01).
|
The enhanced differentiation was also confirmed by the expression changes in C/EBPɛ mRNA and protein in HL-60 cells. C/EBPɛ is a downstream target gene of RAR and a terminal differentiation marker of granulocytes (25). As shown in Fig. 2B, ATRA alone elevated C/EBPɛ mRNA level, but the combined treatment of ATRA with JQ1 synergistically increased the levels of C/EBPɛ mRNA. Similarly, Western blot analysis also revealed a strong upregulation of C/EBPɛ protein after the combined treatment of JQ1 and ATRA, compared to either treatment alone. Taken together, these results suggest that the combination of JQ1 and ATRA has the great potential to induce myeloid differentiation in HL-60 cells.
JQ1 enhances ATRA-induced apoptosis of MV4-11 cells
We next examined whether the synergistic effect of JQ1 and ATRA on suppression of AML cell growth resulted from apoptosis. Annexin V staining revealed that co-treatment with JQ1 and ATRA for 48 h did not have any synergistic or additive effect on apoptosis in HL-60 cells (Fig. 3A). In the case of MV4-11 cells, the combined treatment of JQ1 and ATRA enhanced apoptosis (Fig. 3B). Although the combined effect on apoptosis was not so significant, it may explain, at least in part, its effect on the synergistic growth inhibition.
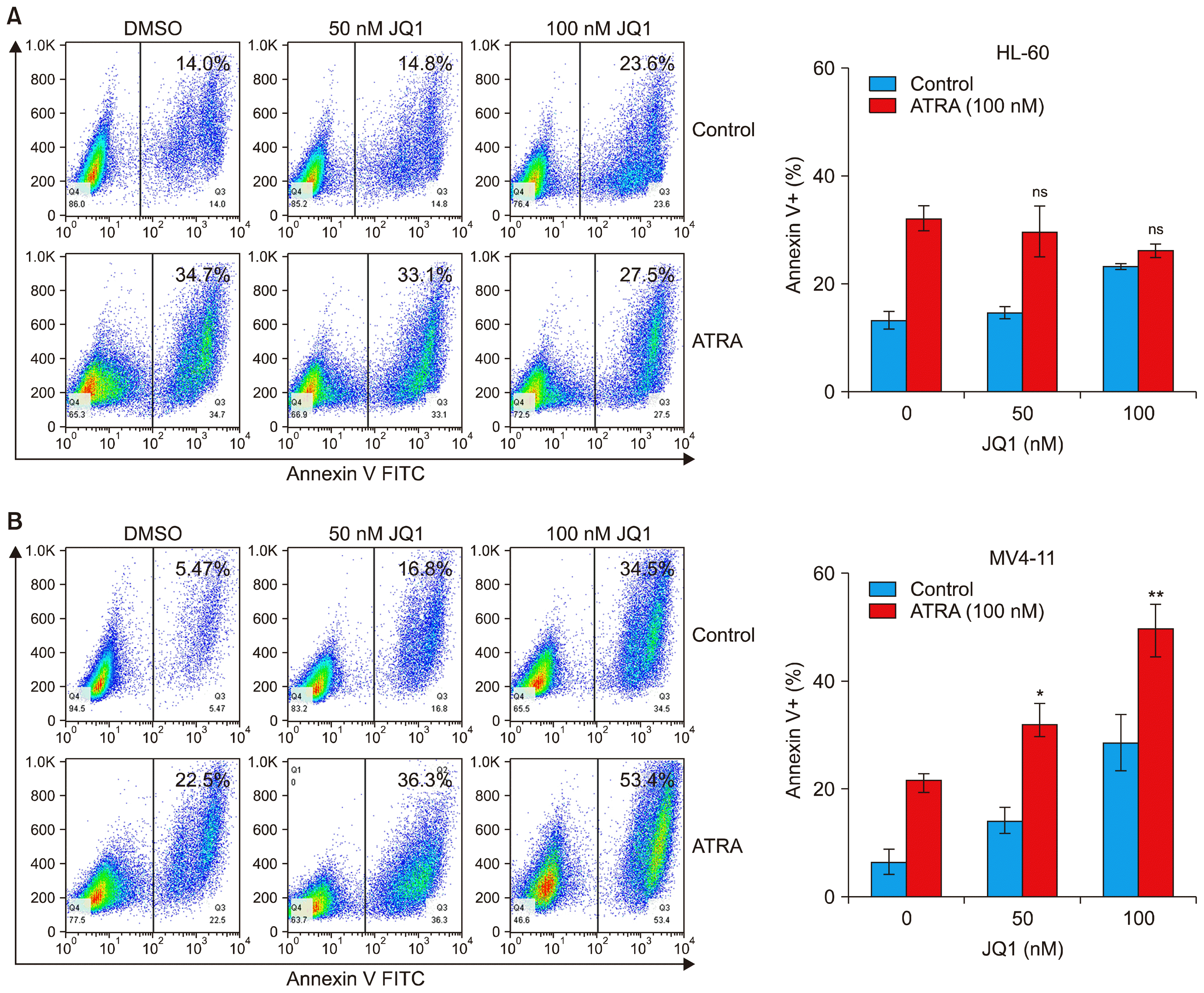 | Fig. 3Effect of combined treatment with JQ1 and ATRA on apoptosis of AML cells. (A) HL-60 cells and (B) MV4-11 cells were treated with the indicated concentrations of JQ1 and/or 100 nM ATRA for 48 h. At the end of treatment, cells were washed with staining buffer and stained with Annexin V-FITC. Annexin-V positive, apoptotic cells was determined by flow cytometry. Numbers in each plot represented the percentage of Annexin-V positive cells. Bar graph showed means±SD of 3 independent experiments. *combination versus ATRA (**p<0.01; *p<0.05; ns, not significant).
|
The combined treatment of ATRA with JQ1 enhances the downregulation of c-Myc and hTERT mRNA in AML cells
To find a mechanism underlying the synergy between JQ1 and ATRA, we investigated expression levels of c-Myc in AML cells, because c-Myc is involved in AML cell proliferation, differentiation, and apoptosis (11, 26). Consistent with previous reports (13, 14), treatment with JQ1 for 24 hours dose-dependently decreased the levels of c-Myc mRNA. (Fig. 4A). ATRA alone also induced downregulation of c-Myc mRNA, and the combined treatment of ATRA with the indicated concentrations (50 nM and 100 nM) of JQ1 caused strong downregulation of c-Myc mRNA, compared to either treatment alone. (Fig. 4A). Corresponding to the decrease in c-Myc mRNA, the combination of JQ1 and ATRA also enhanced the down-regulation of c-Myc protein (Fig. 4B). Thus, the expression levels of c-Myc were correlated with the results of proliferation, differentiation and apoptosis analyses, and may contribute to the synergistic effect of ATRA and JQ1.
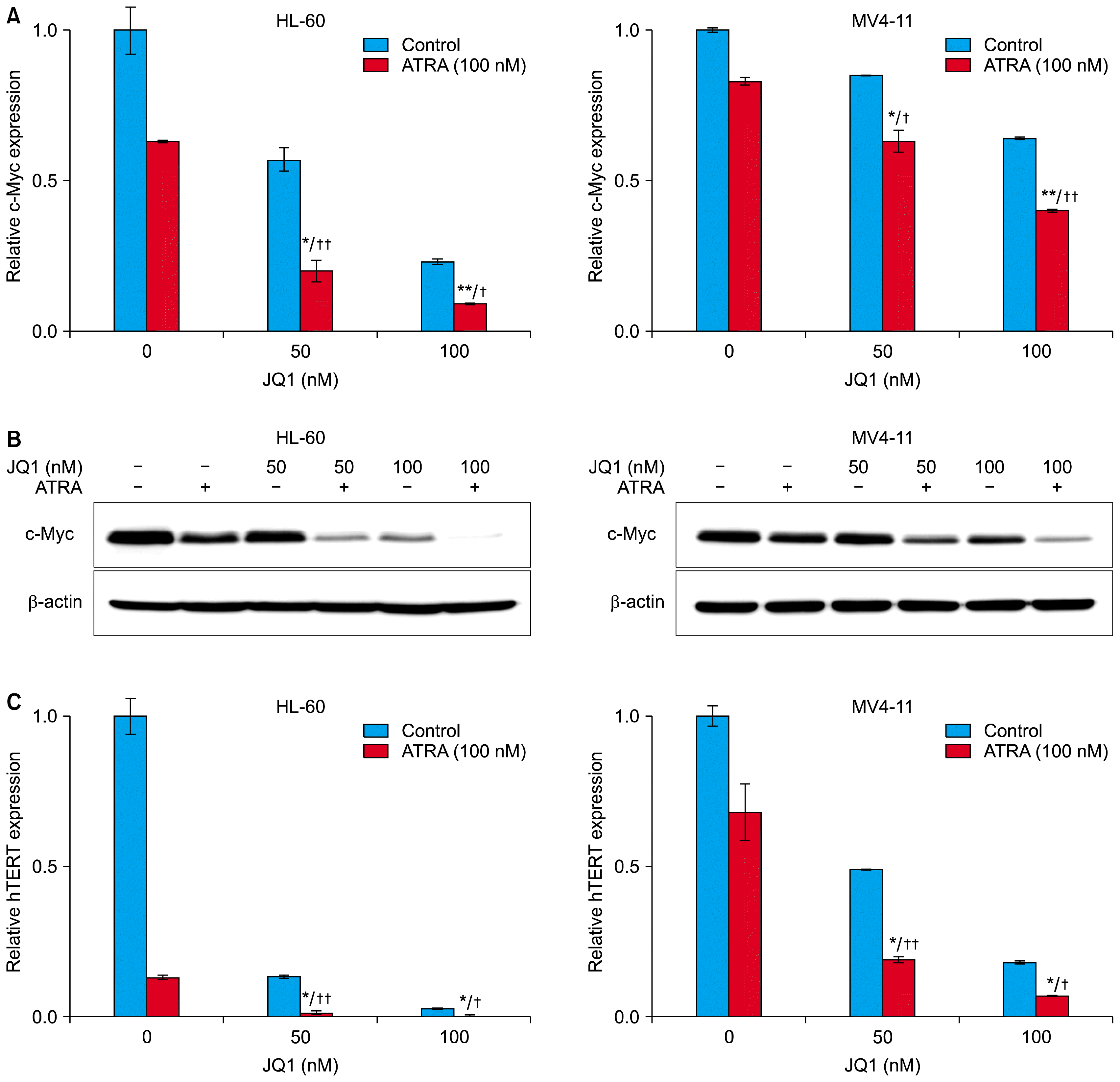 | Fig. 4Effect of JQ1 and ATRA on the expression of c-Myc and hTERT mRNA. HL-60 cells (left) and MV4-11 cells (right) were treated with 100 nM ATRA and/or the indicated concentrations of JQ1 for 24 h. At the end of treatment, cells were harvest and analyzed in the following experiments. (A) The mRNA levels of c-Myc were determined by real-time PCR. The relative expression of c-Myc mRNA was normalized to GAPDH and was reported as a percentage of untreated cells. (B) The protein levels of c-Myc were measured by Western blot analysis using 20 μg protein. Similar results were obtained in three independent experiments. (C) The mRNA levels of hTERT were determined by real-time PCR. The relative expression of hTERT mRNA was normalized to GAPDH and was reported as a percentage of untreated cells. *combination versus ATRA (**p<0.01; *p<0.05); †combination versus JQ1 (††p<0.01; †p<0.05).
|
We next investigated the combined effect of ATRA and JQ1 on mRNA expression of hTERT, a direct target of c-Myc (27). Consistent with previous reports, treatment with 100 nM ATRA for 24 hours decreased the levels of hTERT mRNA in HL-60 cells (Fig. 4C, left). Treatment with indicated concentrations (50 nM and 100 nM) of JQ1 also downregulated hTERT mRNA, but the combined treatment of ATRA with JQ1 caused strong downregulation of hTERT mRNA, compared to either treatment alone in HL-60 cells (Fig. 4C, left). These effects were also observed in MV4-11 cell, although to a lesser extent (Fig. 4C, right). However, hTERT mRNA expression was much lower when cells were treated in the same way for longer time period (48 h, data not shown). These results suggest that the levels of hTERT mRNA are correlated with anti-proliferative synergism between JQ1 and ATRA.
Go to :
Discussion
APL is the only malignancy in which differentiation therapy using ATRA in combination with chemotherapy has resulted in cure (28, 29). However, AML, with exception of APL, fails to respond to the differentiation therapy. Because treatment of ATRA alone is not enough for complete eradication of AML cells, many efforts have focused on the search of effective agents in combination with ATRA (7–9). In this study, we showed that JQ1 could improve the ATRA responsiveness in non-APL AML cells. To our knowledge, it is the first time to show that JQ1 can enhance ATRA responsiveness in non-APL AML cells.
We observed that JQ1 and ATRA synergistically inhibited cell proliferation in both AML cell lines, HL-60 and MV4-11. The synergistic growth inhibitions resulted from different biological responses between two AML cell lines, myeloid differentiation in HL-60 cells and apoptosis in MV4-11 cells.
For the molecular mechanisms underlying the synergy in the combined treatment of JQ1 and ATRA, we focused on c-Myc expression. Inhibition of c-Myc has been shown to induce differentiation and apoptosis in AML cells (11, 30, 31). In this study, we found that the combined treatment of JQ1 and ATRA resulted in a greater decrease of c-Myc than either treatment alone. The greater decrease of c-Myc was correlated with more growth inhibition, differentiation and apoptosis in AML cells. These results indicate that c-Myc inhibition may contribute to the synergistic effect of ATRA and JQ1. Inhibition of BRD4 by JQ1 has been shown to block transcriptional elongation of c-myc gene (15, 16). ATRA is also reported to block c-Myc mRNA elongation (32). Therefore, the double modulation of c-Myc expression induced by these two agents may contribute to the observed synergy in AML cells.
We also examined the expression of hTERT after the combined treatment of JQ1 and ATRA. hTERT expression is directly regulated by c-Myc transcription factor (27) and is the major mechanism to regulate telomerase activity (33). Telomerase is overexpressed in most AML cells and its inhibition eliminates AML cells (34, 35). In this regards, the synergistic effect of JQ1 and ATRA on the downregulation of hTERT expression may be the key to eradication of AML cells.
In this work, we assessed myeloid differentiation in HL-60 cells by CD11b and C/EBPɛ expression. CD11b expression has been used in many studies as myeloid differentiation marker (8, 14, 30, 31). And C/EBPɛ is a downstream target gene of RAR and a terminal differentiation marker of granulocytes (25). We found that JQ1 markedly enhanced ATRA-induced differentiation of HL-60 cells. Although ATRA alone did up-regulate CD11b and C/EBPɛ, we speculated that such increase might not be enough to induce complete maturation but only combined treatment resulting in significant up-regulation of CD11b and C/EBPɛ could speed up terminal differentiation. Recently, c-Myc has been shown to bind RARα and repress ATRA-target genes in AML cells (12). JQ1 is likely to enhance ATRA-induced differentiation in HL-60 cells by relieving the repressive activity of c-Myc on RARα.
On the other hand, the combination and either agent alone had no effect on myeloid differentiation in MV4-11 cells. Instead, consistent with the previous reports (7, 9), ATRA acted as not a differentiation inducing agent but apoptosis inducer in MV4-11 cells that express FLT3-ITD mutation. Combined treatment of ATRA with JQ1 enhanced apoptosis in MV4-11 cells, compared to either treatment alone. However, the combined effect on apoptosis was additive or weakly synergistic, so mechanisms other than apoptosis might be involved in synergistic growth inhibition.
The combined treatment of JQ1 and ATRA enhanced differentiation in HL-60 cells and apoptosis in MV4-11 cells. Inhibition of c-Myc can induce differentiation or apoptosis depending on cell types (11, 26). This may explain the different biological effects observed in HL-60 and MV4-11 cells for the combination of JQ1 and ATRA.
To date, many efforts have focused on the search of agent that can enhance ATRA-induced differentiation in non-APL AML cells (8, 36–38). In this regard, the enhancing effect of JQ1 in ATRA-induced apoptosis shows another aspect of the combinatorial effect against AML. Therefore, we surmised that the combination might broaden the scope of its potential use in AML.
In conclusion, our results show that the combination of JQ1 and ATRA have enhanced therapeutic potential for AML. The combined treatment of JQ1 and ATRA resulted in greater depletion of c-Myc and hTERT expression. These results may explain the synergistic anti-leukemic effects of the combination of JQ1 and ATRA. Although further clinical and mechanistic studies are needed for this combination treatment, our findings presented here underscore their potential use in the treatment of AML.
Go to :
 XML Download
XML Download