

 PDF
PDF Citation
Citation Print
Print


Introduction
Terminally differentiated somatic cells are typically specialized for specific functions and appear to have lost the ability to self-renew due to the inactivation of the pluripotency gene regulatory network (1). Undifferentiated somatic cells, however, often retain pluripotency and are assumed to contribute to tissue homeostasis and regeneration (2). The local microenvironment or niche provides ideal conditions for maintaining stem cell characteristics and behavior (3). Despite the critical role of niches in maintaining stem cell function and dysfunction, most of the current stem cell knowledge has been obtained based on isolating stem cells from their microenvironment through enzymatic methods (4–9). The effect of enzymatic digestion may become more evident when stem cells reside within complex extracellular microstructures like heart valves. For instance, the pulmonary valve (PV) consists of valve endothelial cells (VECs) and interstitial cells (VICs) distributed within a specialized extracellular matrix (ECM). The reversible transdifferentiation of VECs to mesenchymal phenotypes shows the plasticity of a subset of VECs in their response to growth factors and suggests the presence of progenitor cells within the valve leaflets (10, 11). The valve progenitor cells may play a critical role in renewing and repairing the valve tissue during normal homeostasis and disease conditions, however; understanding their biological characteristics remains incomplete due to unavailability of an appropriate cell isolation method. This study introduces a non-enzymatic method for isolation and expansion of the valve tissue stem cells and investigates the phenotype and behavior of the cells in vitro.
Materials and Methods
Tissue collection
Donor hearts were obtained from 26 adult female pigs under post-necropsy tissue collection guidelines at the Stanford Veterinary Service Center. Pulmonary valves were immediately excised and washed three times in cold phosphate-buffered saline (PBS). For histological analysis, one cusp of each pulmonary valve (including small parts of pulmonary artery and right ventricle) was used (n=10 leaflets) and the other two leaflets were dissected for primary tissue culture (n=52 leaflets).
Primary tissue culture
The pulmonary valve leaflet was excised from the ventriculoarterial junction of the cusp and washed multiple times in cold PBS. The leaflet tissue was minced into small pieces and placed in tissue culture petri dishes (Corning, NY, USA) or plastic chamber slides (NuncTM Lab-TekTM, CA, USA). The fragments of the leaflet were covered by DMEM culture media (Gibco®, CA, USA) containing 5% NCS (Gibco®, CA, USA) and incubated at 37°C with 5% CO2. The outgrowth of adherent and non-adherent cells over the subsequent 12 weeks was evaluated.
Histology
The dissected cusp was fixed in 10% buffered formalin (Fisher Scientific, Fair Lawn, NJ, USA) for 24 hours and then embedded in paraffin. Serial cross-sections of the leaflets (5 micrometer) were prepared for Russell-Movat pentachrome staining (American Master Tech Scientific, Inc., Lodi, CA, USA) and immunohistological analysis.
Antibodies
Mouse monoclonal primary antibodies were applied to identify fibronectin (BD Biosciences, CA, USA), versican (Asher laboratory, Department of Physiology, University of Cambridge, UK), HSP47 (Abcam, MA, USA), vimentin (Abcam, MA, USA), CD31 (Abcam, MA, USA), eNos (Biomol, HH, Germany), α-SMA (Dako, CA, USA), PCNA (Santa Cruz Biotechnology, CA, USA), SSEA-1 (Abcam, MA, USA), SSEA-2 (Abcam, MA, USA), TRA-1-60 (Millipore, CA, USA), and TRA-1-81 (Millipore, CA, USA). Rabbit polyclonal primary antibodies were used to recognize von Willebrand factor (vWF) and c-Kit (Dako, CA, USA), Nanog (Abcam, MA, USA), Oct-4 and vascular endothelial growth factor receptor-1 (VEGFR-1), and VEGFR-2 (Santa Cruz Biotechnology, CA, USA).
Immunohistochemistry (IHC)
The valve tissue sections from 10 animals were deparaffinized and rehydrated to PBS by immersion in a descending ethanol series. The sections were permeabilized in 0.2% Triton X-100 in PBS for 30 minutes and then washed twice with PBS. For immunofluorescent staining only, autofluorescence was reduced by washing the sections 3 times in fresh 1 mg/ml sodium borohydride (Sigma-Aldrich, CA, USA), followed by three 10-min washes in PBS. Antigen retrieval was performed using a citrate buffer method (BD Biosciences, CA, USA) at 90~100°C for 10 minutes to unmask the antigenic epitopes, after which the staining container was placed at room temperature for 30 minutes to allow the slides to cool. The sections then were subjected to three 10-min washes in PBS, covered with a blocking buffer (10% blocking serum in PBS), and incubated for 1 hour in a humidified chamber at room temperature. The primary antibodies were prepared according to the respective vendor’s instructions and applied to the sections, and the sections were incubated overnight at 4°C. On the second day, the sections were subjected to three 10-min washes in PBS before being incubated in the dark at room temperature with fluorescent tag-conjugated (Invitrogen, CA, USA) or biotinylated (Vector Laboratories, CA, USA) secondary antibodies for 1 hour. The sections then were subjected to three 10-min washes in PBS. For fluorescent imaging, the sections were counterstained with DAPI and covered with ProLong Gold antifade mounting solution (ThermoFisher Scientifics, CA, USA), and the signal was evaluated using an upright fluorescence microscope (Leica Microsystem, CA, USA). For DAB staining, sections were incubated in the appropriate dilution of Avidin-Biotin complex (Vector laboratories, CA, USA) for 30 minutes followed by three 10-minute washes with PBS. Staining was developed by using DAB substrate (Dako, CA, USA), with counterstaining using hematoxylin (Poly Scientific, NY, USA). The sections were dehydrated in an ascending ethanol series, cleared in xylene, and covered with coverslips using mounting solution. The micrographs were recorded using a multispectral imaging system (CRi, MA, USA). The staining was repeated 3 times for each antibody.
Immunocytochemistry (ICC)
Cells were washed twice with prewarmed PBS and fixed in 4% formaldehyde solution for 10 minutes. Cells were permeabilized in 0.2% Triton X-100 in PBS for 30 minutes and then washed twice with PBS. Next, cells were incubated with an appropriate blocking serum for 1 hour at room temperature. Primary antibodies were appropriately prepared for double staining and added to cells, and the mixture was incubated overnight at 4°C. The staining was completed on the second day as described above for IHC. In total, the leaflet-derived cells from 5 animals were used for ICC and the staining was repeated 2 times for each antibody.
Viability/Cytotoxicity assay
The viability of the cellular structures derived from the in vitro culture of pulmonary valve leaflets was investigated using the LIVE/DEAD® Assay (Molecular Probes, Invitrogen life Science, California). Live cells were demonstrated by staining with green-fluorescent calcein-AM indicating intracellular esterase activity whereas necrotic and apoptotic cells were stained with red-fluorescent ethidium homodimer-1 indicating damage to the cellular membrane.
Cytoskeleton staining
Non-adherent and adherent cells were separately washed with PBS and fixed in 4% formaldehyde solution for 10 minutes. Actin staining was performed using Alexa FluorTM 488 phalloidin (Invitrogen, CA, USA) according to the vendor’s instructions. The adherent cells were viewed using a Leica DMI 6000B microscope (Leica Microsystem, CA, USA) and the images were recorded using a Leica DFC 360 FX camera (Leica Microsystem, CA, USA). The 3D structures of the non-adherent colonies were visualized using a Leica TCS LSI microscope (Leica Microsystem, CA, USA).
Fluorescent live-cell tracking
The supernatant medium of the 3- to 4-week-old primary culture was collected and replaced with fresh medium. The collected supernatant was centrifuged and the resulting supernatant was aspirated and discarded; the remaining cell content (pelleted non-adherent cells) was resuspended in either green or red CellTrackerTM fluorescent dye solution (Invitrogen, CA, USA) for 30 minutes under standard growth conditions. Then, cells were centrifuged and the supernatant was aspirated and discarded. To remove the excess fluorescent dye, cells were washed three times with PBS, with each wash consisting of resuspension in PBS, centrifugation, and aspiration/discarding of the supernatant. The labeled cells were added back to the primary culture and the mixture was incubated under standard growth conditions. The interaction between green- and red-labeled cells in the primary culture was recorded every 4 minutes for 10 days using live fluorescence and time-lapse imaging.
Types of culture media
Non-adherent cells were collected from the primary culture as described above. Cells were distributed into 6 separate culture dishes and cultured in each of the various media. DMEM containing 5% NCS was used as a reference medium. Endothelial Growth Medium and Mesenchymal Stem Cell Growth Medium were obtained from Lonza, MD, USA. Smooth Muscle Cell Growth Medium was obtained from Cell Application, CA, USA. RESGRO Culture Medium was obtained from Millipore, CA, USA. Morula-like structures and non-adherent mature colonies were aspirated using a pipet tip and subcultured solely in DMEM containing 5% NCS. The growth and phenotype of the new cultures were investigated using light microscopy as described above (for primary tissue culture).
Results
Expression of stem cell markers in porcine PV leaflets
To further study the characteristics of valve stem cells, we initially characterized the ECM and cellular components of adult porcine PV using immunohistological staining (Supplementary Fig. 1). Fibronectin was the major ECM expressed on both the ventricular and arterial sides of PVs, whereas the expression of versican was localized primarily within the spongiosa and fibrosa. HSP-47 (a collagen marker) and vimentin were expressed predominantly by VICs and VECs covering the ventricular side of the leaflets. Expression of endothelial cell markers was observed in VECs; only a small population of VICs expressed the myofibroblast marker. To investigate the pluripotency of valve cells, the serial sections of PV tissue were separately stained for stem cell markers. The expression of c-Kit and Oct-4 was mainly observed within and below the VECs (Fig. 1A and 1B). A subset of cells expressed α-smooth muscle actin (α-SMA) on the ventricular side, just below the VECs (Fig. 1C). Positive staining for proliferating cell nuclear antigen (PCNA) was detected in VECs and VICs (Fig. 1D). Other embryonic cell markers such as stage-specific embryonic antigen-1 (SSEA-1), stage-specific embryonic antigen-4 (SSEA-4), keratan sulfate antigen TRA-1-60 (Tra-1-60), and keratan sulfate antigen Tra-1-81 (Tra-1-81) were not detectable in the PV leaflets (Fig. 1E~H). Co-expression of the stem cell marker c-Kit and the endothelial cell marker e-Nos was observed in VECs (Fig. 1I) and in some of the sub-endothelial cells adjacent to the subset of α-SMA-positive cells (Fig. 1J). These results suggest the dual character of VECs and identified colonies of cells expressing α-SMA within the PV ventricular side.
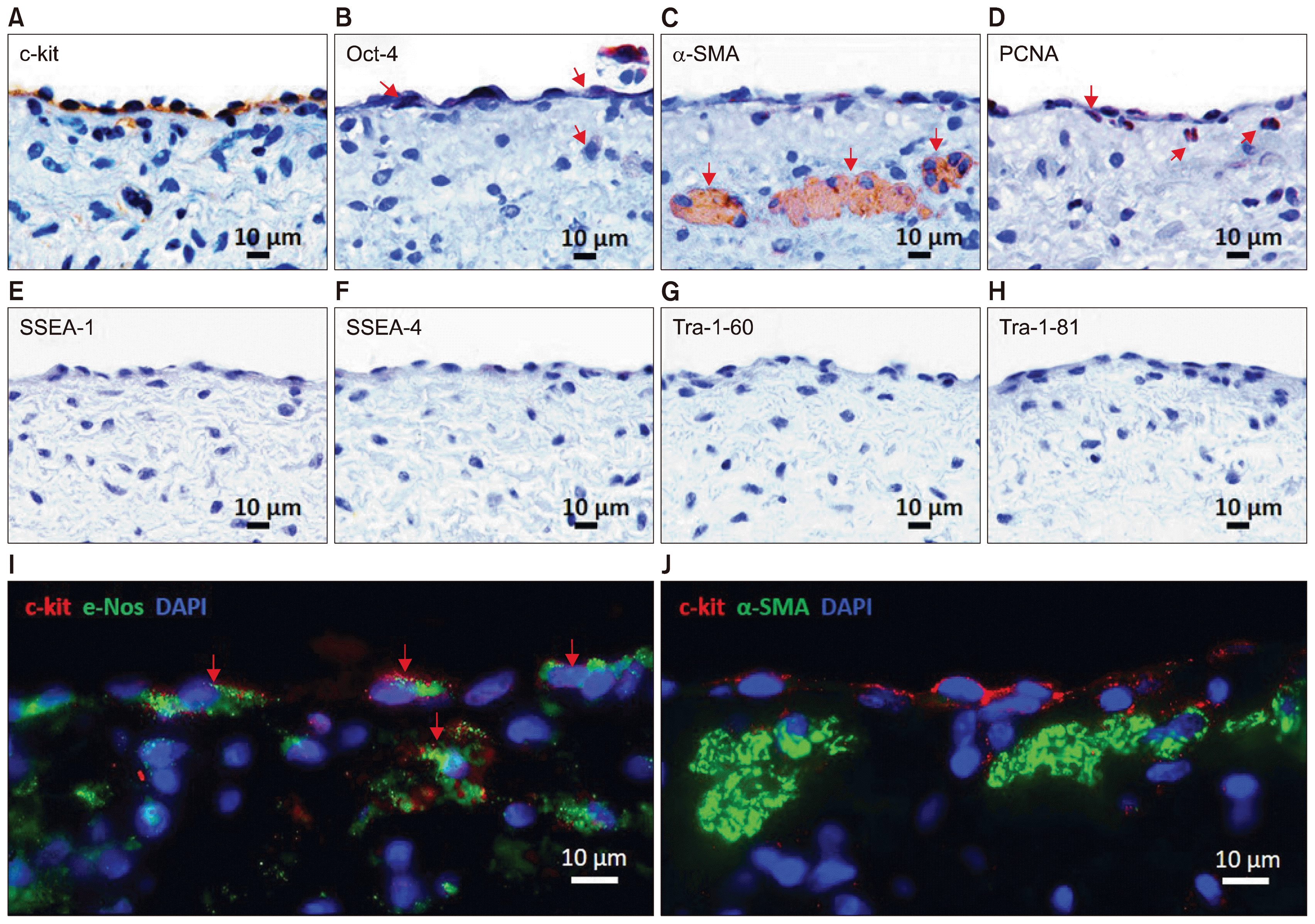
Fig. 1
Expression and colocalization of stem cell markers in the leaflet (A~J). IHC staining showed the presence of c-Kit+ (A; arrows) and rare Oct-4+ cells (B; arrows) within VECs. Expression of α-SMA was observed in a subset of multinucleated cells (C; arrows) below the endothelial surface of the ventricular side. The positive staining for PCNA showed the proliferation potential of VECs and VICs (D; arrows). The expression of pluripotent stem cell markers SSEA-1 (E), SSEA-4 (F), Tra-1-60 (G), and Tra-1-81 (H) was not detected in the representative tissue sections. Co-expression of c-Kit (red) and e-Nos (green) in VECs (I; arrows) was confirmed by immunofluorescent staining. In addition, a subset of c-Kit+ cells (red) was found in the subendothelial region directly adjacent to the α-SMA+ multinucleated cells (J; green). DAPI (blue) was used for counterstaining of the nuclei.
![]()
Formation of embryonic-like structures in vitro
To isolate the pluripotent cells, 52 PV leaflets were harvested from 26 animals (2 leaflets from each animal). The minced leaflets were cultured separately under the same conditions and characteristics of the cell outgrowths were monitored for 12 weeks (Fig. 2). Migration of fibroblast-like adherent and round motile, non-adherent cells from the cultured leaflets was observed during the first week (Fig. 2A). The formation of multinucleated cells was followed by the retraction of adjacent adherent cells, creating a niche area (Fig. 2B) for recruiting and interacting with the non-adherent cells (Fig. 2C and 2D). The development of bubble-like cellular structures was initially observed during the second week (Fig. 2E). These structures mimicked the phenotypes of oocytes; 2-, 3-, and 4-cell embryos; and morulae (Fig. 2F~H). Aggregation of non-adherent cells into the niche and the morula-like structures resulted in the formation of dense spheres and blastula-like colonies (Fig. 2I~K). Incorporation of the adjacent niche areas resulted in the generation of a larger syncytium (Fig. 2L) in which interconnecting bundles were formed between the poles of the nuclei, suggesting active networking between the colonies. The structure of the cells and colonies were examined using nuclear and cytoplasmic staining. In each oocyte-like structure, a single nucleus was localized in the periphery and formed a ring-like structure (Fig. 2M); however, the nuclear morphology of the other bubble-like structures was ambiguous due to the recruitment of the non-adherent cells around the bubble-like membrane (Fig. 2N). A small number of non-adherent cells were observed to penetrate in and out of the bubble-like membrane without rupturing the membrane (data not shown). The viability of the multinucleated cells and colonies was confirmed by intracellular esterase activity in the living cells, distinguishing them from the apoptotic and necrotic cells (Supplementary Fig. 2). The blastula-like structures each consisted of a hollow sphere of individual cells with the capacity to harbor non-adherent cells (Fig. 2O and Supplementary Video 1). The F-actin cytoskeleton was diffusely distributed in the cytoplasm of the fused multinucleated cell (Fig. 2P), indicating the role of actin filaments in expanding the size of the niche area.

Fig. 2
Formation of diverse cellular structures from the leaflet during 12 weeks of tissue culture (A~P). Outgrowth of adherent fibroblast-like cells started a few hours after placing the leaflet tissue in the culture; these fibroblast-like cells stabilized the attachment of the leaflet tissue to the culture dish and provided a feeder layer for non-adherent cells (A). Retraction of the confluent feeder layer created a space (B) for formation of multinucleated syncytium-like structures (C) and a niche area (D). By the second week, bubble-like structures of differing sizes and shapes were detected (E). The phenotype of these structures was similar to those of oocytes, 2-cell embryos, 3-cell embryos (F), 4-cell embryos (G), and morulae (H). Formation of large dense colonies and hollow spheres occurred in the confluent cultures between week 3 and 12 (I). The dense colonies were formed either by recruiting the non-adherent cells to the morula-like structures (J) or by the retraction of the mature niche area (K). Interconnecting bundles were generated between adjacent niche syncytia (L, arrow), a process that often was followed by unification of the two structures to form a larger colony. Phalloidin staining (green) and DAPI nuclear counterstaining (blue) showed the structure of the embryonic-like cells and colonies. Oocyte-like cells exhibited a ring-shaped structure with an elongated and peripheral nucleus (M). Multiple nuclei were located on the periphery of 2-cell-embryo-like structure (N), indicating the possibility that other non-adherent cells would be arranged around these structures. Two-dimensional imaging of one blastula-like structure showed a round aggregation of cells with distinct borders between the cells (O). The unification of two adjacent niche areas was promoted by cytoplasmic fusion (P).
![]()
Expression of pluripotent markers in the PV leaflet-derived cells
The expression profiles of the cell populations were characterized using immunofluorescence staining, and the co-expression of multiple markers was investigated. The non-adherent cells expressed c-Kit and connected with the underlying surface and with other cells through the adhesion molecule versican (Fig. 3A). The expression of versican increased in the c-Kit+ multinucleated cell aggregates, where the protein presumably promoted cell adhesion. The cavity of the blastula-like structure was partially covered by c-Kit+ cells and was filled with versican (Fig. 3B). The pluripotent markers Nanog and Oct-4 were expressed by newly formed multinucleated cells in the niche, which were surrounded by α-SMA+ adherent cells (Fig. 3C and 3D). The spontaneous differentiation of Oct-4+ cells to e-Nos+ endothelial cells was observed only in mature colonies (Fig. 3D). These results raised questions regarding the origin of the niche pluripotent cells.
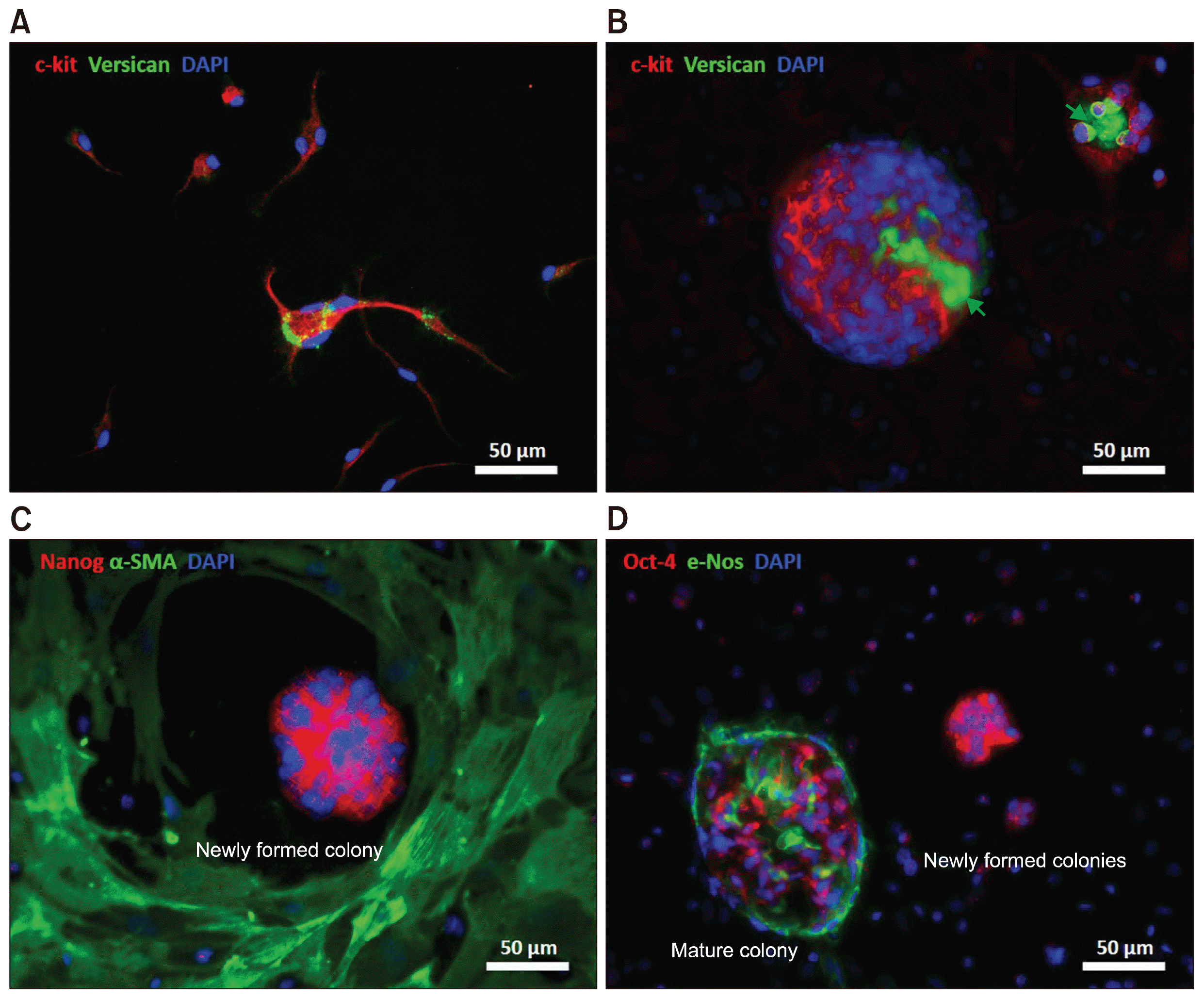
Fig. 3
Characterization of different types of multinucleated cells in a culture of the leaflet (A~D). Expression of c-Kit was detected in the majority of non-adherent mononuclear and multinuclear cells (A, B; red). Versican was expressed by multinucleated cells and found in the inner cavity of blastula-like colonies, suggesting a potential role for versican in cell aggregation and tissue regeneration (A, B; green arrows). Myofibroblast characteristics of the adherent cells surrounding the niche area were identified by expression of α-SMA (C; green). Expression of pluripotent markers Nanog and Oct-4 were detected in the newly formed niche areas before progressively decreasing during development of multinucleated colonies (C, D; red). Expression of the endothelial cell marker e-Nos in mature colonies showed spontaneous differentiation of pluripotent stem cells into endothelial cells (D; green).
![]()
Role of cell fusion in the niche formation
To investigate the contribution of c-Kit+ cells in niche formation, the non-adherent cells were collected from the 3- to 4-week-old primary culture and processed for use in a fluorescent tracing study. The cells were divided into two groups and each group was incubated either with green or red fluorescent dye for 30 minutes and then washed. Aliquots of both the red-labeled and green-labeled cells were returned to each culture and the dynamics of the culture were monitored for 10 days using live fluorescence and time-lapse imaging. A subset of the labeled cells (both red and green) adhered to the culture dish and either initiated niche formation or transformed to adherent fibroblast-like cells (Fig. 4A~D). Other subsets of the labeled cells were integrated into the existing multinucleated syncytium (Fig. 4E~H), bubble-like structures (Fig. 4I~L), and blastula-like colonies (Fig. 4M~P) that had formed before addition of the labeled cells. A sub-group of the labeled cells remained highly motile and non-adherent, and these cells were involved in partial and complete cytoplasmic cell fusion. During the partial cell fusion, the cytoplasmic content of the donor was partially delivered to the recipient cell; however, this exchange reduced motility of the recipient cell and recruited the recipient’s enzymatic reactions (as evidenced by the known chemistry of the CellTracker dyes) (12, 13) to produce the cytoplasmic content of only the donor cell or both donor and recipient cells (Supplementary Video 2). Although this assay showed the role of cytoplasmic exchange in alteration of cellular behavior and function in PV cells, the delivery of nuclear contents could not be ruled out during this process. The complete cell fusion events were detected primarily in the niche area; the non-adherent cells were the predominant source of the fusion and detachment of new cells into and from the niche area, respectively (Supplementary Video 2). A reversible consolidation of cell contents and formation of rosette shape fragments was observed during the partial and complete fusions, explaining the different sizes and phenotypes of the non-adherent cells.
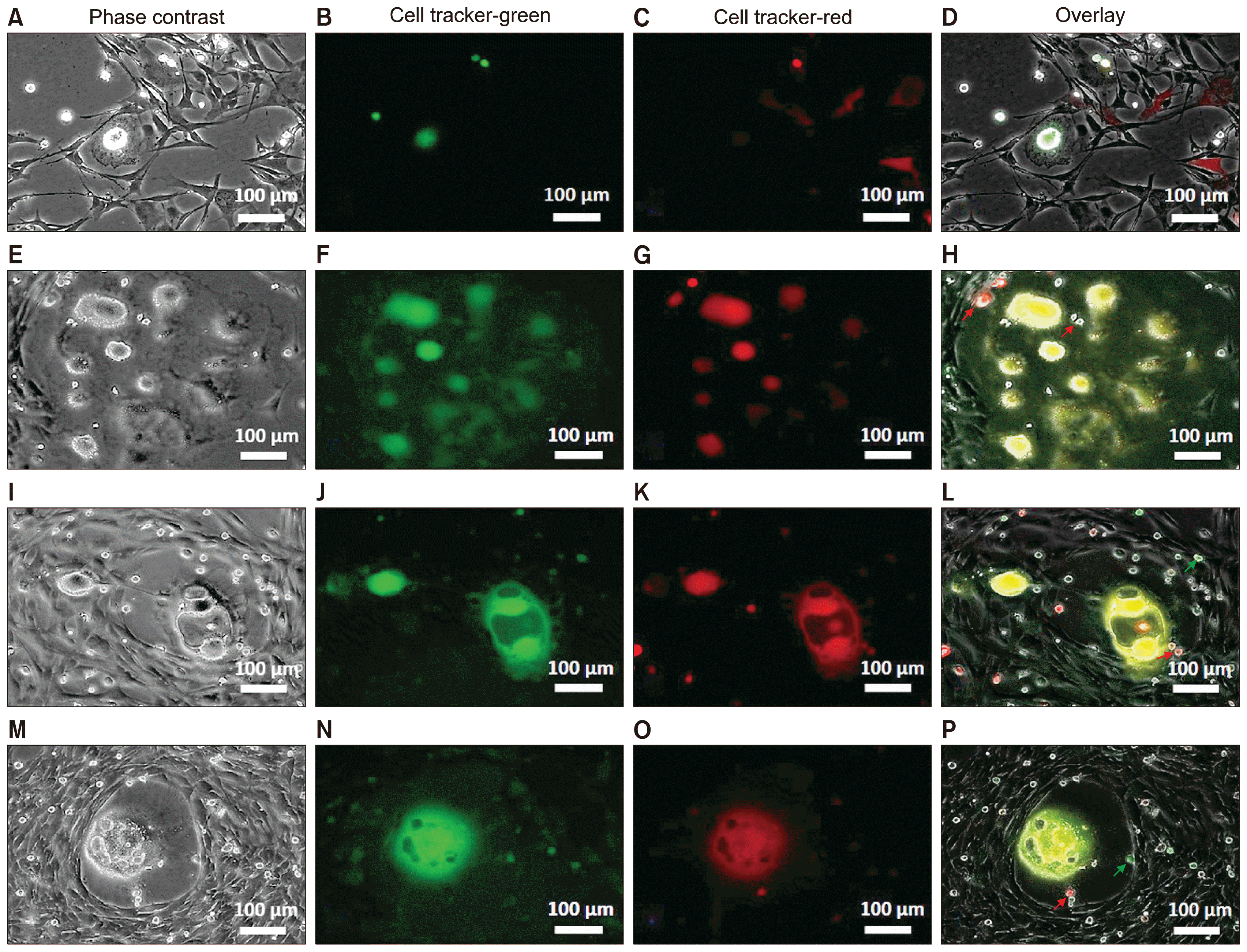
Fig. 4
Tracing fusion of non-adherent cells using fluorescent dyes of two different colors (A~P). Non-adherent cells were collected and stained (separately) with green or red cell-tracker fluorescent dyes; cells of each color were returned to the originating culture. During the first few days, some of the labeled cells attached to the dish, whereas some other cells remained non-adherent (A~D). Cytoplasmic fusion of green and red labeled cells (yellow in the overlay) was observed in syncytium-like structures (E~H), bubble-like structures (I~L), and multinucleated niche areas (M~P). Migration of labeled cells into the existing and newly formed niche areas was detected (H, L, P; arrows).
![]()
Role of culture media in alteration of the non-adherent cell phenotype
To examine the effect of culture media on differentiation of non-adherent cells, the non-adherent cells were collected and cultured in each of five different culture media (see Methods). When Dulbecco’s Modified Eagle’s medium (DMEM) (supplemented with 5% Newborn Calf Serum [NCS]) was used for the primary tissue culture of the leaflets, the collected non-adherent cells reproduced the characteristics of the primary culture by 2~3 weeks after re-seeding (Fig. 5A, F, L). In Endothelial Growth Medium (EGM) or Smooth Muscle Cell Growth Medium (SMCGM), the collected non-adherent cells exhibited phenotypes consistent with differentiation into endothelial (Fig. 5B, G, M) or smooth muscle (Fig. 5C, H, N) cells, respectively. In Mesenchymal Stem Cell Growth Medium (MSCGM), non-adherent cells proliferated and differentiated into adherent cells, which in turn increased the cell density and colony formation over the course of the first week of culturing (Fig. 5D, I, O). In RESGRO medium, the formation of niche area and proliferation of non-adherent cells dominated (Fig. 5E, J, K, P, Q). These results demonstrated that non-adherent cells yielded cells of various phenotypes in the individual media, confirming the proliferation and differentiation potential of this class of cells. Notably, RESGRO appears to be a suitable medium for maintenance and proliferation of the pluripotent non-adherent cells; in contrast, a standard medium like DMEM can be used as a cost-effective medium for primary culture of the leaflets and passaging of the non-adherent cells.
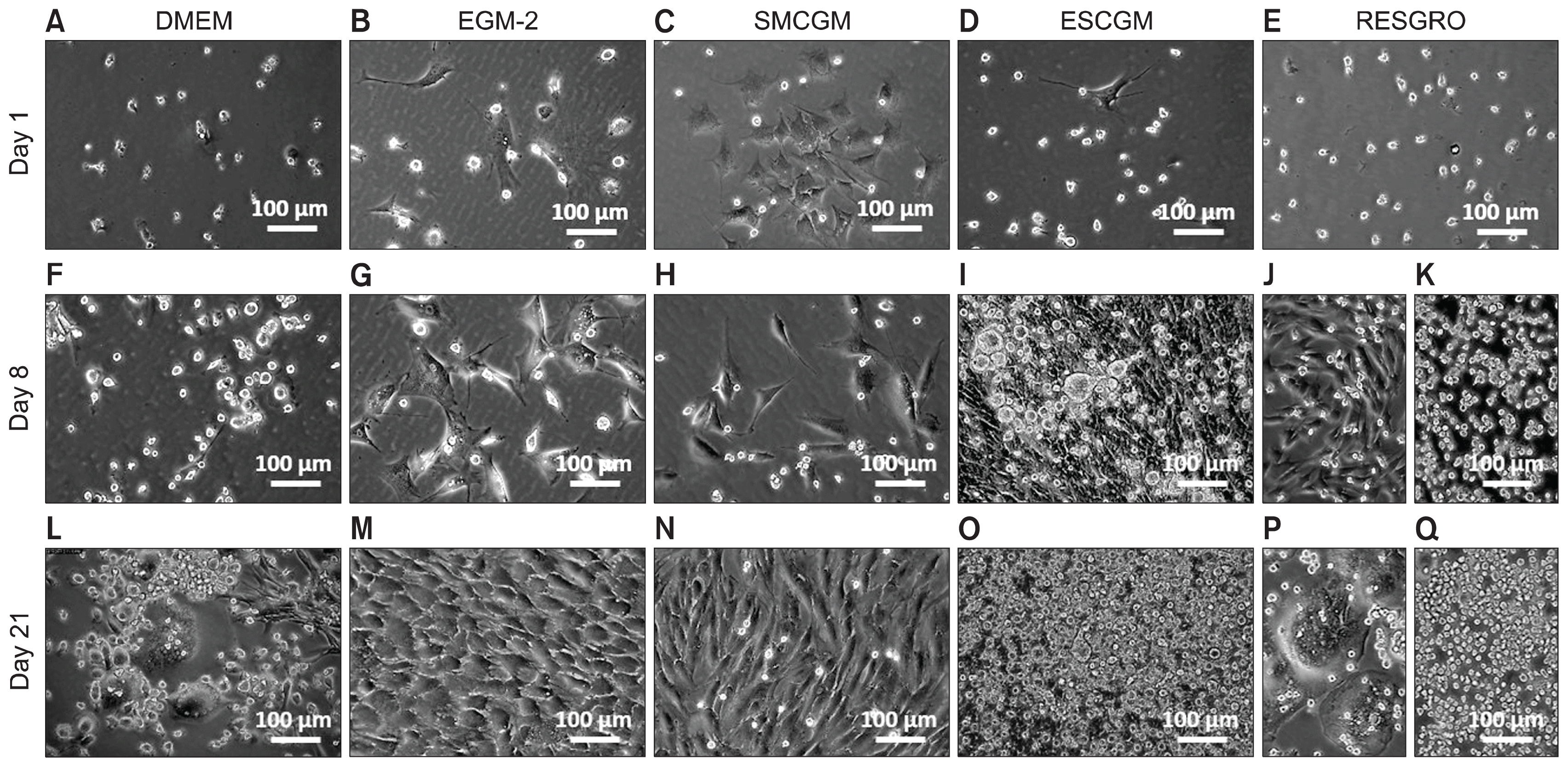
Fig. 5
Differentiation of the non-adherent cells in growth culture media. Non-adherent cells were collected from the primary culture of leaflets and grown in DMEM (A), EGM-2 (B), SMCGM (C), MSCGM (D), or RESGRO (E) medium. During the first week, non-adherent cells exhibited low levels of proliferation in DMEM medium (F) and changes in phenotype (e.g., by adhering to culture dishes) in EGM-2 or SMCGM medium (G, H). The proliferation and adhesion rates were strongly increased in MSCGM (I) or RESGRO (J, K) medium. Formation of multinucleated colonies was widely detected in DMEM or RESGRO medium (L, P) after 3 weeks, whereas proliferation of non-adherent cells and the maturation of the colonies was the evident in MSCGM or RESGRO medium (O, Q). Differentiation of non-adherent cells into cells with endothelial or myofibroblast phenotype was detected in EGM-2 or SMCGM culture, respectively (M, N).
![]()
The differentiation potential of the isolated morula-like structures and mature colonies was investigated. In DMEM, the regression of morula-like structures was observed within a few days after isolation (Supplementary Fig. 3A, B). This result suggested the essential role of both the feeder-adherent and non-adherent cells in the maintenance of these structures; however, the potential for further development of these structures is still debatable. In contrast, the mature colonies generated predominantly fibroblast-like cells, subsequently yielding dense colonies with a low number of non-adherent cells (Supplementary Fig. 3C, D). This observation indicated that differentiated cells constituted the major subpopulation of cells within the colony.
Discussion
Although the results of this study showed the presence of stem cells and their ability to form multinucleated niche areas in the in vitro culture, the origin of these cells is speculative. We identified four main cell types within native PV leaflets: VECs co-expressing endothelial cell markers and c-Kit, VICs producing extracellular matrices, sub-endothelial cells expressing α-SMA, and pluripotent cells expressing Oct-4. In in vitro culture, migration of VICs from the leaflet was critical for the formation of the fibroblast-like feeder layer and attachment of tissue fragments to the culture dish. In vivo, the differentiation of VICs into myofibroblast cells occurs during valve development, remodeling, and disease, and this differentiation is characterized by the increased expression of α-SMA and a change in the phenotype of the VICs (14, 15). Differentiation of VICs to myofibroblast cells occurred in our in vitro culture and provided a feeder-like layer for growing and maintaining the embryonic-like structures and multinucleated syncytia. Although the formation of embryonic-like stem cells and the ability of these cells to express embryonic stem cell transcription factors Oct-4 and Nanog was demonstrated, the involvement of the PV tissue-resident Oct-4+ cells in the development of the pluripotent colonies is questionable. Oct-4+ cells have been identified in adult healthy and tumor tissues by some investigators (16–23); however, other laboratories have reported that Oct-4+ cells are not required for somatic cell self-renewal and tumor formation (24, 25). The latter studies identified Oct-4 pseudogenes in hematopoietic stem cells that were not involved in pluripotency (24, 25), and showed the regeneration capacity of mouse intestinal epithelium, bone marrow, hair follicle, brain, and liver tissues after Oct-4 gene ablation (26). In the present study, the ability of embryonic-like colonies to generate c-Kit+ stem cells was associated with the expression of Oct-4 and Nanog, suggesting the involvement of these two embryonic transcription factors in the colonies’ self-renewal process. In the mature colonies, however, the expression of Oct-4 was reduced, and regenerating colonies produced only endothelial- and fibroblast-like cells.
Formation of oocyte-like cells from fetal pig skin (27) and mouse fibrosarcoma L929 cells (28) has been reported by other investigators. Although the expression of meiosis gene SCP-3 was shown in oocyte-like cells, the majority of these cells did not enter meiosis properly (28, 29). Single-clone analysis of the L929 cells suggested two possible explanations for the spontaneous formation of embryonic-like structures: (1) the presence of existing germ cells in the pool of fibroblast cells, or (2) the re-acquisition of germline characteristics due to mutation or epigenetic factors (28). In the present study, formation of embryonic-like cells was observed predominantly in the first 4~6 weeks of primary culture, suggesting activation of pre-existing germ-like cells already present within the valve tissue. Nevertheless, the regression of isolated morula-like structures in the fresh medium indicated a potential role for the microenvironment in providing the required factors for further development of the germ-like cells. In addition, spontaneous cell fusion may induce the state of pluripotency and reprogram the valve somatic cells in vitro. Our results confirmed the ability of the non-adherent c-Kit+ cells to form multinucleated structures, vacuolated colonies, and endothelial and interstitial cells, even after multiple passages. The differentiation of c-Kit+ cells into endothelial and interstitial cells in vitro suggests the potential contribution of these cells to valve repair and self-renewal in vivo. We speculate that in vivo alteration of epigenetic factors may cause an imbalance in the homeostasis of the c-Kit+ cells, changing the phenotype and function of the cells, promoting cell fusion, and leading to pathological changes in valve tissues.

 XML Download
XML Download