

 PDF
PDF Citation
Citation Print
Print


Introduction
Mesenchymal stem cells (MSCs) are a multipotent class of stem cell with potential as a cell-based therapy for both autoimmune and neurological diseases (1). While MSCs can be derived from a variety of tissues, one of the most common and readily available sources of MSCs for clinical applications is the bone marrow (2). Bone marrow derived MSCs (BM-MSCs) are a non-hematopoietic type of stem cell that can be collected and expanded in-vitro from both human donors and experimental animal models (2).
BM-MSCs possess a remarkable capacity to modulate the responses of different immune and neural cell types (3). For example, BM-MSCs have strong immunosuppressive functionality (4), and can inhibit the proliferation and activation of a range of immune cells including T-cells, B-cells, and macrophages (1, 5). These immunosuppressive capabilities appear to be mediated in large part by the secretion of a broad spectrum of anti-inflammatory chemokines and cytokines by BM-MSCs (6). In addition to their immunomodulatory capabilities BM-MSCs are also neuroprotective and may influence the development of distinct classes of neural cells in the CNS. For example, BM-MSCs as well as conditioned medium from BM-MSCs biases neural stem cell fate away from an astroglial fate and promotes neural stem cell differentiation into oligodendrocytes and/or neurons (7, 8). In addition, BM-MSCs and BM-MSC conditioned medium enhances differentiation of oligodendrocyte precursor cells (OPCs) into mature oligodendrocytes (9). The effects of BM-MSCs on neural cells also appears to be mediated by secreted factors, including growth factors like hepatocyte growth factor (HGF) (10).
Due to their immunosuppressive and neuromodulatory properties, BM-MSCs are considered to be a potentially powerful cellular therapy for inflammatory and neurodegenerative diseases of the CNS such as multiple sclerosis (MS) (11). Experimental autoimmune encephalomyelitis (EAE) is a commonly used model of MS that results in a predictable onset of functional deficits that correlate with immune mediated CNS demyelination (12). When BM-MSCs are transplanted intravenously into EAE they rapidly halt disease progression and improve motor recovery and function (8, 13, 14). Although the mechanisms mediating this functional improvement are not well defined, transplanted BM-MSCs appear to suppress the immune response as well as the activation and migration of peripheral immune cells into the CNS of EAE mice (8, 13). In addition, BM-MSCs attenuate demyelination and possibly promote remyelination and axonal survival (8, 13, 15). The therapeutic benefits of BM-MSCs in EAE appear to be mediated by their secretion of anti-inflammatory and pro-regenerative factors (10, 16).
Based on their success in treating animal models of MS like EAE, BM-MSCs have moved into clinical trials for MS patients. The majority of these trials use autologous MSCs (derived from MS patients) (17), in contrast to most previous studies in EAE animal models, which used MSCs from healthy human donors or healthy animals (“naïve MSCs”) (8, 13, 14). New evidence suggests however that inflammatory disease can alter the therapeutic functionality of BM-MSCs. In a recent study, BM-MSCs derived from EAE mice and MS patients were shown to have reduced therapeutic efficacy compared to naïve MSCs in their ability to ameliorate disease progression in EAE (18). The MSCs derived from donors with ongoing disease secreted higher levels of many pro-inflammatory cytokines, and demonstrated less immunosuppressive potential in-vitro (18).
Defining how diseases like MS affect the functionality of BM-MSCs is crucial in evaluating whether autologous MSCs represent a viable strategy for treating MS and other inflammatory diseases. The current study was undertaken to gain insight into how disease affects BM-MSCs at a transcriptional level though RNA-Sequencing experiments to compare gene expression profiles between naïve MSCs and MSCs derived from EAE mice (EAE-MSCs). Alternations in the expression levels of a large number of genes between EAE-MSCs and naïve MSCs were detected using this approach, with bioinformatic analysis revealing significant perturbations in pathways related to inflammation and immune cell regulation as well as myelination and neural cell fate. Such changes in gene expression in EAE-MSCs correlated with important functional consequences, as conditioned medium from EAE-MSCs failed to promote oligodendrogenesis in vitro and instead favored astroglial expansion. Collectively, this data underscores how inflammatory diseases like EAE dramatically alter BM-MSCs at a transcriptional and functional level, raising concern about the efficacy of using autologous MSCs as a cellular therapy for MS.
Go to : 
Materials and Methods
EAE induction and scoring
All animal experiments were performed according to protocols approved by Case Western Reserve University School of Medicine’s IACUC with adherence to the NIH Guide for the Care and Use of Laboratory Animals.
EAE was induced in 10 week-old female C57BL/6 mice (Jackson Laboratory: 000664) using Hooke Labs MOG35–55 EAE Induction kits according to the manufacturer’s protocol. Briefly, mice were immunized via subcutaneous injection of 200 μl of MOG35–55 peptide in complete Freund’s adjuvant. Pertussis toxin (250 ng) was injected intraperitoneally at 2 and 24 hours post immunization. Animals began showing signs of paralysis 9~11 days post immunization, and were graded by blinded observers according to a previously described clinical index (10): 1=limp tail, 2=hind limb weakness, 3=plegia of one limb, 4=plegia of two limbs, 5=moribund or dead.
MSC culture and RNA isolation
Mesenchymal stem cells were isolated and cultured from the bone marrow of MOG35–55 – induced EAE mice at 14 days (peak EAE-MSCs) or 28 days (chronic EAE-MSCs) after immunization, with the animals having a clinical score of 4. Naïve MSCs were cultured from non-immunized, age-matched C57BL/6 mice. Growth medium for all cultures consisted of α-MEM with GLUTAMAX (Gibco) supplemented with 10% MSC-qualified fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco).
BM-MSCs were isolated as previously described (18). Briefly, bone marrow from the tibias and fibulas was collected by flushing the central canal of the bone with a 26 ×g syringe containing fresh growth medium. Bone marrow cells were then seeded in P75 flasks (Corning) at a concentration of 2×105 cells/cm2 and grown at 37°C with 5% CO2. Flasks were washed twice with media 48 hours later to remove non-adherent cells, and the medium changed every 2~3 days. Cells were passaged using 0.25% Trypsin/EDTA (Gibco) for 2 minutes at 37°C and re-plated in P75 flasks at a concentration of 1×104 cells/cm2.
At the third passage (21 to 28 days in-vitro), 1×106 MSCs were collected for RNA extraction using RNeasy Mini kits (Qiagen) according to the manufacturer’s protocol. An independent culture preparation consisting of MSCs derived from 2~3 mice was used for each RNA sample. Conditioned medium (CM) was likewise collected from passage 3 naïve and EAE-MSCs between 21 and 25 days in vitro, with unconditioned medium collected as a control. All CM samples were filtered through a 0.22 μm filter, centrifuged at 10,000×g for 10 minutes, then frozen at −80°C. Conditioned medium was collected from a different culture preparation for each experiment.
RNA Sequencing
RNA libraries were prepared using TruSeq Stranded Total RNA Library Prep kits with Ribo-Zero Gold according to the manufacturer’s protocol. Samples were analyzed for whole-transcriptome analysis using Illumina HiSeq 2500 v2 (RAPID RUN), with a read length of 1×50 bp for gene expression analysis. RNA-Seq data can be accessed through NCBI’s Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) with submission number SUB3074064.
Mapping of reads and differential expression analysis
Mapping of reads and differential expression analysis was done in BaseSpace using the RNA Express Legacy application (version 1.0.0). Reads were mapped to the Musmusculus – UCSC (build mm10) reference genome. Criteria for differentially expressed genes included a fold change of 1.5 or greater, q-value ≤0.05, and a normalized mean expression count ≥10.
Gene ontology and pathway analysis
Gene ontology analysis of the top 2,000 differentially expressed genes (ranked by greatest magnitude fold change) was performed using DAVID (version 6.7). Further gene pathway analysis of all differentially expressed genes was performed using MetaCore. Heat maps were constructed using OriginLab Pro (version 2017) with genes mapped according to their respective z-score.
Neural cell culture and immunocytochemistry
Dissociated neural cell cultures were prepared as previously described (8). Briefly, spinal cords from P3 C57/B6 mice were dissected manually and chemically dissociated in 0.1% Trypsin/EDTA for 20 minutes and passed through a 30 μm cell strainer to generate single cell suspension. These cells were then plated onto poly-L-lysine coated glass coverslips and grown in neural cell culture medium (DMEM supplemented with 10% FBS and 1% antibiotic).
To test the effects of MSC CM of neural cell development and differentiation, cells were cultured in medium that contained 50% neural cell culture media and 50% MSC CM or unconditioned medium (controls) for 4 days. Coverslips were fixed for 10 minutes in 4% paraformaldehyde and labeled for respective cell markers using a previously published protocol (8). Antibodies used include: rat anti-MBP (Abcam: ab7349 at 1:100), rabbit anti-GFAP (Millipore: ab5804 at 1:800), and rabbit anti-NG2 (Millipore: ab5320 at 1:200). To quantify the percentage of cells labeled with each respective marker, 6 fields per coverslip were counted, with 2–3 coverslips per experiment over 2 independent experiments.
Statistical analysis
For RNA sequencing and expression experiments, statistical analysis was performed via the RNA express app in BaseSpace, with q-values (multiplicity adjusted p-values) of ≤0.05 considered statistically significant. For cell counting experiments, statistical tests were performed using GraphPad Prism 6, with Dunnett’s multiple comparison tests performed post-hoc for one-way ANOVAs and p-values of ≤0.05 considered statistically significant.
Go to :
Results
RNA-Seq reveals disease-related gene expression changes in BM-MSCs in EAE
RNA-Seq analysis was conducted on MSCs derived from the bone marrow of MOG35–55-induced EAE mice at two distinct phases of disease: at 14 days post induction during the peak of the disease (“peak EAE-MSCs”) and at 28 days post induction during the chronic phase of the disease (“chronic EAE-MSCs”). These specific time points were selected based on a previous study showing BM-MSCs derived from MOG-induced EAE mice at these stages of disease have a diminished capacity to suppress inflammation and ameliorate EAE when compared to MSCs derived from non-diseased donors (18). To identify transcriptional changes underlying the reduced therapeutic functionality in EAE-MSCs, gene expression profiles of both peak and chronic EAE-MSCs were compared to expression profiles of naïve MSCs derived from healthy, sex-matched littermate controls.
To ensure that control MSCs were appropriately age-matched to their respective disease counterparts, different cultures of naïve MSCs were derived from healthy littermates at the time of peak EAE and two weeks later for chronic EAE. At least three biological replicates were used for gene expression comparisons and analysis. A different primary culture preparation, consisting of 1×106 BM-MSCs expanded for 3 passages (21~28 days in-vitro) and derived from 2~3 mice, was used for each replicate.
RNA-Seq analysis revealed widespread changes in gene expression patterns in EAE-MSCs that correlated with disease progression. Differentially expressed genes (DEGs) between EAE-MSCs and naïve MSCs were defined by a magnitude fold change of 1.5 or greater, q-value ≤0.05, and a normalized mean expression count ≥10. Using these criteria, RNA-Seq analysis identified 2,337 DEGs between peak EAE-MSCs and naïve MSCs (Fig. 1A). Chronic EAE-MSCs had more pronounced changes in gene expression, with 5, 837 DEGs between chronic EAE-MSCs and naïve MSCs (Fig. 1A). The magnitude of individual gene expression changes in chronic EAE-MSCs also tended to be higher than the magnitude of gene expression changes in peak EAE-MSCs (Fig. 1A). For example, when comparing chronic EAE-MSCs to naïve MSCs, 34% of DEGs showed a fold change of 3 or greater, whereas only 12% of DEGs showed a fold change of 3 or greater when comparing peak EAE-MSCs to naïve MSCs. Unsupervised hierarchical clustering showed peak EAE-MSC and chronic EAE-MSC replicates clustered together independently of naïve MSC replicates (Fig. 1B). These data indicate that both peak EAE-MSCs and chronic EAE-MSCs have distinguishable and reproducible changes in gene expression patterns compared to naïve MSCs.
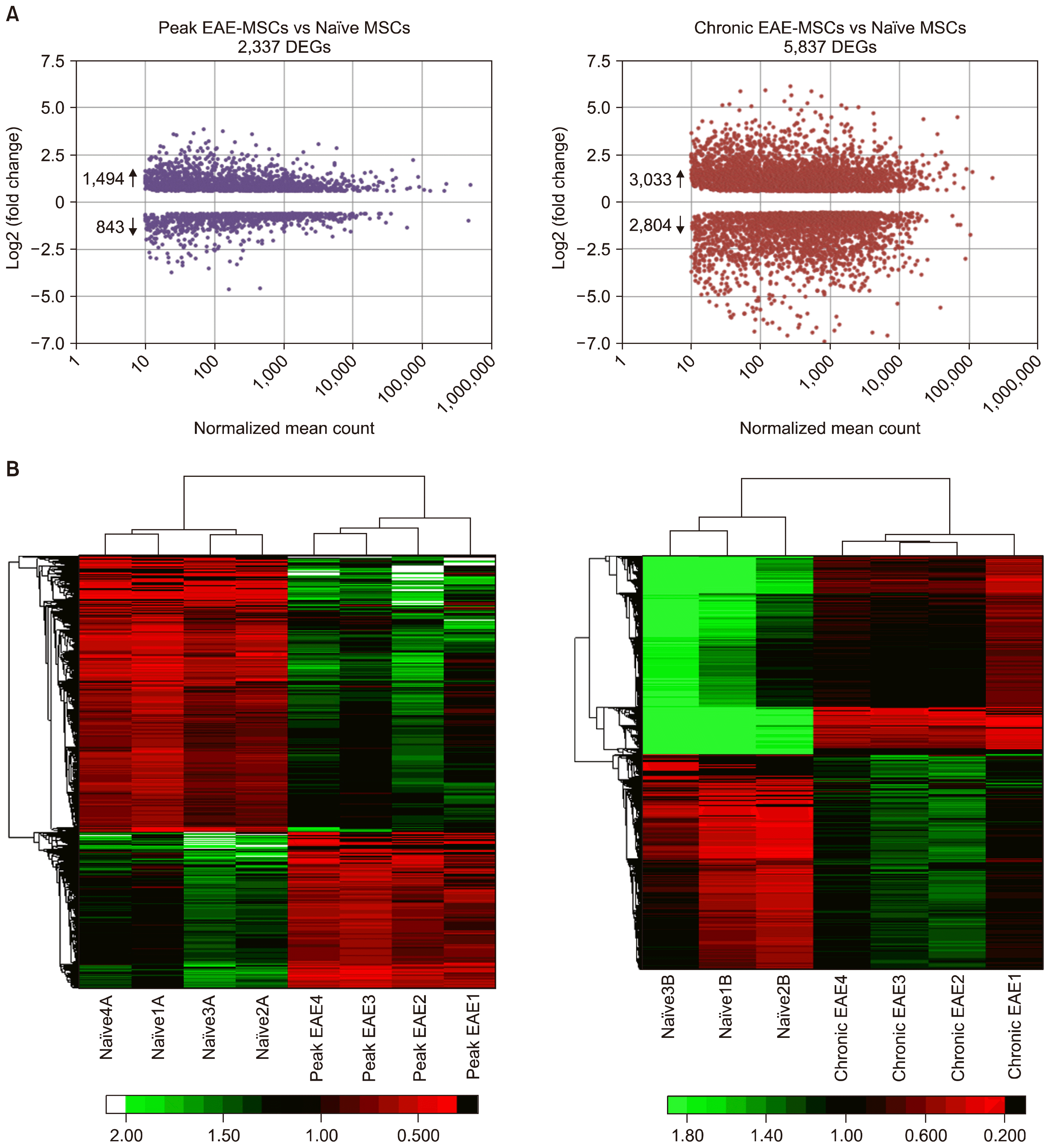 | Fig. 1RNA-Seq analysis of gene expression changes in BM-MSCs in EAE. (A) Volcano plots depicting differentially expressed genes (DEGs) measured by RNA-Seq analysis of peak EAE-MSCs versus naïve MSCs (left) or chronic EAE-MSCs versus naïve MSCs (right). DEGs were defined by a magnitude fold change of 1.5 or greater, q-value ≤0.05, and a normalized mean expression count ≥10. (B) Unsupervised hierarchical clustering of samples using Pearson correlation with average linkage, along with corresponding dendrogram and heatmap of DEGs. Peak EAE-MSC and chronic EAE-MSC replicates cluster together independently of naïve MSC replicates.
|
EAE-MSCs up-regulate genes linked to inflammation and immune activation
The immunomodulatory and regenerative capacity of BM-MSCs in models of MS is derived in large part from their ability to secrete a wide array of diverse chemokines, cytokines, and trophic factors that may act on a act on a variety of cellular targets, including adaptive and innate immune cells involved in inflammation (6). To identify cellular and biological processes associated with the gene expression changes in EAE-MSCs, gene ontology (GO) enrichment analysis was performed on the DEGs found between naïve and EAE MSCs.
Gene ontology analysis suggested many DEGs are secreted factors or linked to the secretory pathway. For example, comparison of the distribution of DEGs amongst different cellular compartments revealed a large number of DEGs for both peak and chronic EAE-MSCs were associated with vesicles or the endoplasmic reticulum (ER) and Golgi apparatus (Fig. 2). Out of the top five specific cellular compartments DEGs mapped to, vesicles and the ER/Golgi apparatus were third and fourth respectively for both peak EAE-MSCs and chronic EAE-MSCs. The only compartments with a higher level of associated DEGs were the nucleus and cell membrane (Fig. 2).
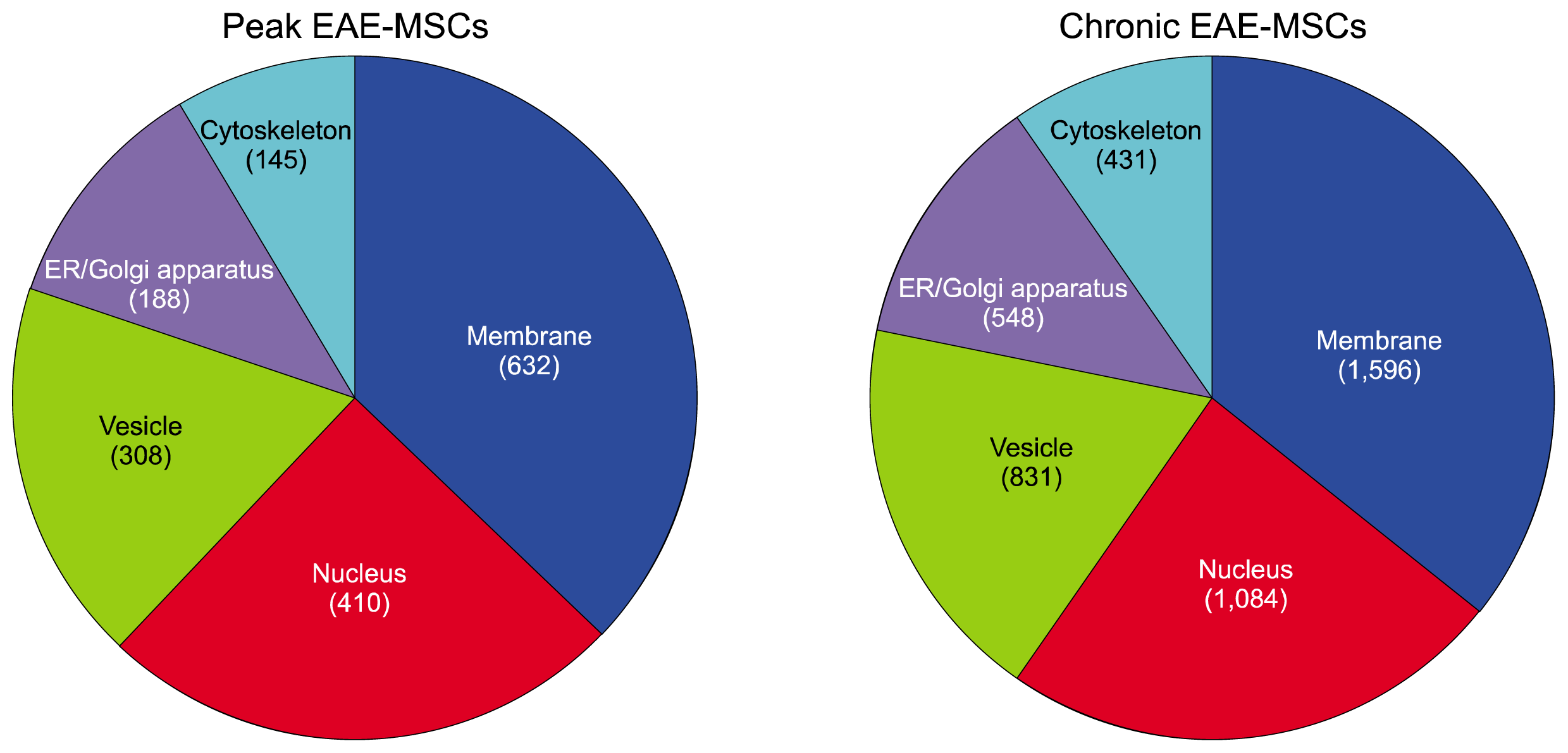
Gene ontology enrichment analysis revealed many DEGs in EAE-MSCs were linked to immune activation and inflammation. For example, analysis of the DEGs for both peak EAE-MSCs and chronic EAE-MSCs showed that the third most enriched term (ranked by p-value) was “immune response” (Fig. 3A). Many of the GO terms associated with DEGs for both peak and chronic EAE-MSCs related to aspects of cell-mediated immunity or inflammatory response, including the terms “lymphocyte mediated immunity, “leukocyte chemotaxis”, “positive regulation of immune response” and “activation of immune response.” (Fig. 3A).
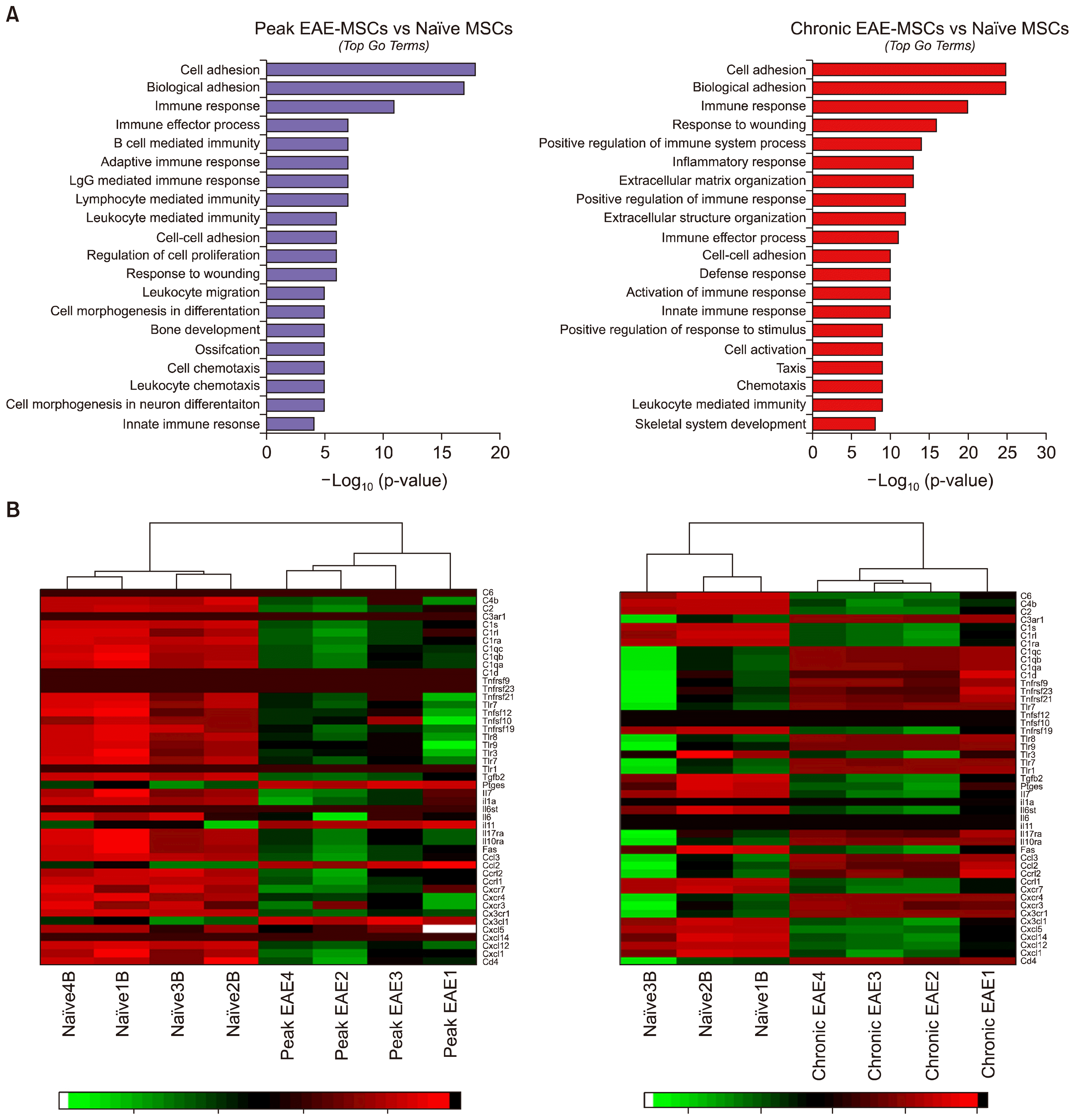 | Fig. 3EAE-MSCs show changes in genes associated with inflammation and immune activation. (A) Top 20 gene ontology terms (ranked by p-value) for DEGs found between peak EAE-MSCs and naïve MSCs (left) or chronic EAE-MSCs and naïve MSCs (right). Most GO terms relate to immune processes, particularly immune activation and immune response. (B) Heatmap showing immune genes differentially expressed between peak EAE-MSCs and naïve MSCs (left) or chronic EAE-MSCs and naïve MSCs (right). Many inflammatory genes, including chemokines, cytokines and elements of the complement system, are up regulated in EAE-MSCs.
|
RNA-Seq analysis revealed an up-regulation of many immune and pro-inflammatory genes in EAE-MSCs compared to naïve MSCs. These included a number of inflammatory chemokines, cytokines, and other factors previously shown to be important in mediating MSC immunomodulation. For example, peak and chronic EAE-MSCs had elevated gene expression of multiple pro-inflammatory chemokines including Cxcl1 and Cxcl5, as well as interleukins important in T-cell activation such as IL-6 (Fig. 3B) (19). This data is consistent with a previous study showing that EAE-MSCs express higher amounts of each of these pro-inflammatory cytokines at the protein level and secrete higher levels of these factors compared to naïve MSCs (18). Previous studies have demonstrated that MSC-induced activation of the complement system after transplantation may reduce MSC viability and compromise their therapeutic efficacy (20). RNA-Seq analysis showed higher gene expression of many complement system factors in EAE-MSCs compared to naïve MSCs, suggesting enhanced complement activation by EAE-MSCs (Fig. 3B). The therapeutic efficacy of BM-MSCs is also influenced by Toll-like receptor (TLR) signaling that can affect the immunomodulatory characteristics of MSCs (21). The expression of many TLR genes was increased in EAE-MSCs compared to naïve MSCs (Fig. 3B), which may contribute to the functional differences between the two cell populations.
Further analysis of DEGs was performed using MetaCore to identify specific biological pathways associated with the gene expression changes observed in EAE-MSCs. MetaCore analysis revealed many DEGs in EAE-MSCs were associated with pathways important in immune cell signaling and immune response. For example, gene expression changes in peak EAE-MSCs and chronic EAE-MSCs were linked to the regulation of T-cell differentiation and activation, and to signaling pathways important in T-cell polarization (Fig. 4A). These results are consistent with previous findings from GO enrichment analysis of the DEGs in EAE-MSCs that found immune and inflammatory processes were significantly over-represented (Fig. 3A). Together the comparative analysis of gene expression between naïve and EAE BM-MSCs reveals an extensive change in immune associated genes. This is consistent with the notion that EAE-MSCs are more pro-inflammatory than naïve MSCs and have reduced immunosuppressive functionality compared to naïve MSCs both in-vitro and in-vivo after transplantation into EAE mice (18).
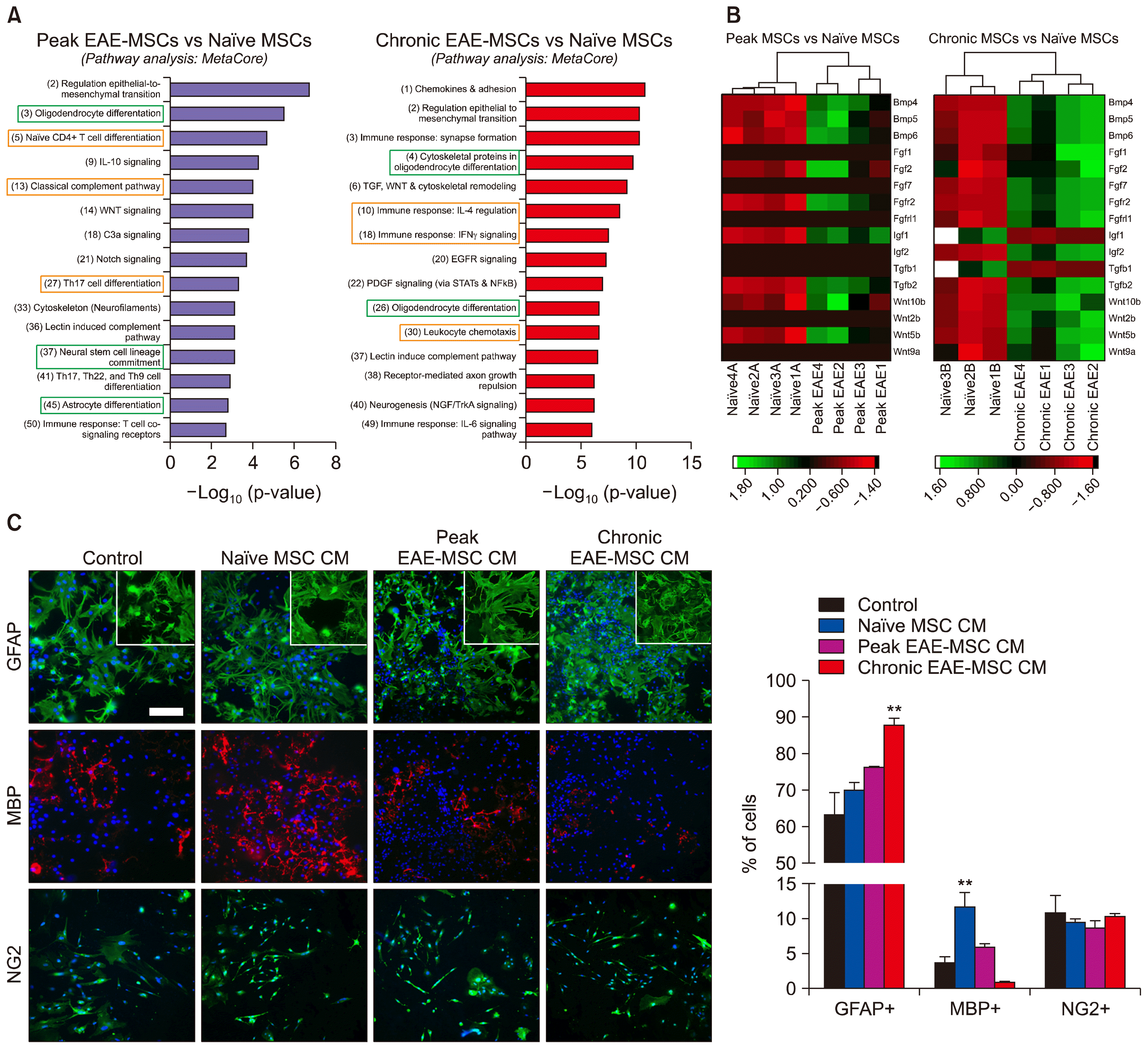 | Fig. 4EAE-MSCs differentially regulate neural cell development and oligodendrocyte formation. (A) Top pathways (ranked by p-value) associated with the DEGs found between peak EAE-MSCs and naïve MSCs (left) or chronic EAE-MSCs and naïve MSCs (right). The number in parenthesis next to the pathway identifies statistical ranking assigned by MetaCore. Notable immune and neural cell pathways have been highlighted in green and red respectively. (B) Heatmap showing expression changes between peak EAE-MSCs and naïve MSCs (left) or chronic EAE-MSCs and naïve MSCs (right) for genes important in regulating neural cell development. (C) Neural cell cultures treated with conditioned medium (CM) from naïve MSCs had significantly higher number of MBP+ oligodendrocytes compared to control cultures treated with unconditioned medium, whereas cultures treated with EAE-MSC CM had a higher number of GFAP+ astrocytes relative to controls and they appear more reactive (insert). Scale bar=30 μm, data shown in graph=mean+SEM, **p<0.01, One-way ANOVA.
|
EAE-MSCs differentially regulate neural cells and oligodendrogenesis
In addition to being immunomodulatory, previous studies have suggested that naïve MSCs have the capacity to influence neural cell fate (7–9). Metacore analysis of the DEGs between EAE-MSCs and naïve MSCs identified changes in pathways important in regulating neural cell development. In both peak and chronic EAE-MSCs, multiple DEGs were associated with “oligodendrocyte differentiation,” and expression changes in peak or chronic EAE-MSCs were linked to “neural stem cell lineage commitment,” “astrocyte differentiation,” and “neurogenesis” (Fig. 4A). Signaling pathways important in neural cell differentiation and development, including WNT, EGFR, and PDGF signaling (22), were also associated with gene expression changes in peak and chronic EAE-MSCs (Fig. 4A).
EAE-MSCs showed changes in gene expression for multiple factors known to regulate neural cell development and oligodendrocyte differentiation. For example, both peak EAE-MSCs and chronic EAE-MSCs up-regulated expression of Fgf and Bmp genes, including Fgf2, Bmp4, and Bmp6 (Fig. 4B). FGF and BMP proteins are important in influencing neural cell fate, as they favor the differentiation of neural stem cells (NSCs) and oligodendrocyte precursor cells (OPCs) into astrocytes versus their differentiation into oligodendrocytes (23, 24). Chronic EAE-MSCs also down regulated expression of genes like Tgfb1 and Igf1, which code for growth factors that promote oligodendrocyte maturation and differentiation (25, 26).
Such changes in gene expression suggest EAE-MSCs may differentially influence neural cell fate compared to naïve MSCs. To test this hypothesis, dissociated cultures of spinal cord neural cells were treated with conditioned medium (CM) from naïve MSCs or EAE-MSCs. Cultures were treated for 4 days (with unconditioned medium used as a control) and the proportion of GFAP+ astrocytes and MBP+ oligodendrocytes was assayed to determine whether factors secreted by EAE-MSCs differentially bias neural cell development.
While cultures grown in CM from naïve MSCs had increased numbers of mature oligodendrocyte compared to controls, cultures grown in CM from chronic EAE-MSCs had reduced numbers of mature oligodendrocytes and increased numbers of astrocytes (Fig. 4C). Parallel studies using CM from Peak EAE-MSCs showed a less significant effect with a slight increase in the number of astrocytes and no reduction in the number of oligodendrocytes (Fig. 4C). In both cultures the presence of EAE-MSC CM increased astrocyte reactivity resulting in elevated expression of GFAP and a more dense network of processes. Changes in the relative number of oligodendrocytes in cultures treated with CM from chronic EAE-MSCs appeared to reflect changes in oligodendrocyte maturation since no significant effect on the number of NG2+ oligodendrocyte precursors was seen compared to controls (Fig. 4C). No changes were seen in the relative number of neurons or microglial cells, which at less than 5% each accounted for a very small percentage of cultured cells (data not shown). These data suggest that the failure of EAE-MSCs to promote recovery in animal models of CNS demyelination may be associated in part with their reduced capacity to promote the development of mature oligodendrocytes.
Changes in gene expression in BM-MSCs during EAE correlate with disease progression
RNA-Seq analysis suggests that gene expression changes in BM-MSCs during EAE evolve with disease progression. For example, the number of DEGs is much higher when comparing chronic EAE-MSCs to naïve MSCs versus comparing peak EAE-MSCs to naïve MSCs. To investigate what genes are differentially expressed between peak and chronic EAE-MSCs, expression profiles of peak EAE-MSCs and chronic EAE-MSCs were compared to each other using previous criteria for DEGs: magnitude fold change of 1.5 or greater, q-value ≤0.05, and a normalized mean expression count ≥10.
RNA-Seq analysis revealed BM-MSCs show continual changes in gene expression patterns throughout EAE progression. There were 3,736 DEGs between peak EAE-MSCs and chronic EAE-MSCs (Fig. 5A). Consistent with comparative studies between naïve and EAE-MSCs, GO analysis identified many of these genes as being associated with immune or inflammatory processes (Fig. 5B). Also, GO analysis suggested many of these DEGs are associated with processes involved in cell uptake and secretion, as “phagocytosis,” “membrane invagination,” and “endocytosis” were among top GO terms (Fig. 5B).
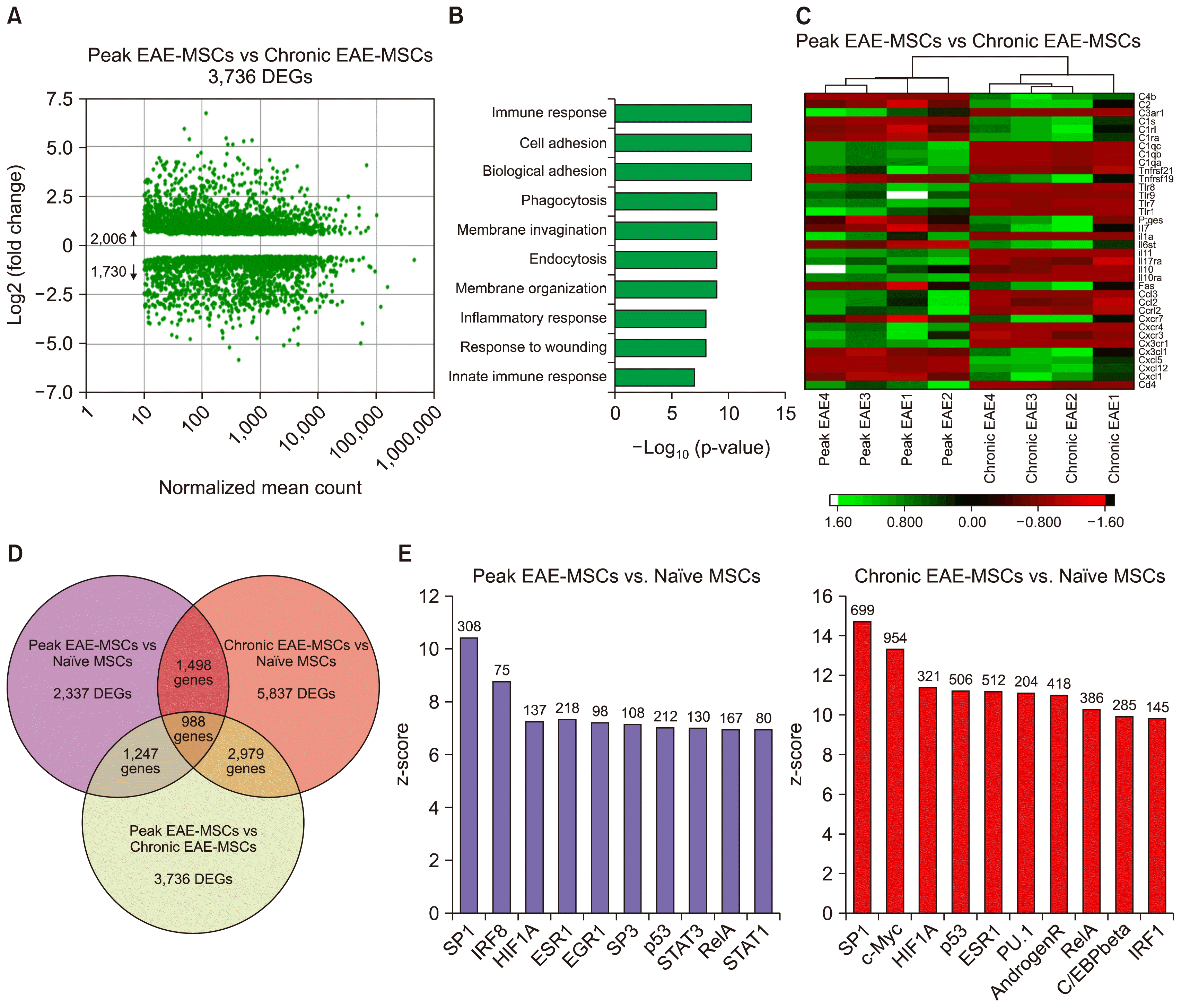 | Fig. 5Changes in gene expression in BM-MSCs throughout EAE. (A) Volcano plot depicting differentially expressed genes (DEGs) measured by RNA-Seq analysis of peak EAE-MSCs versus chronic EAE-MSCs. (B) Top 10 gene ontology (GO) terms ranked by p-value associated with the DEGs found between peak and chronic EAE-MSCs. (C) Heatmap showing immune genes differentially expressed between peak EAE-MSCs and chronic EAE-MSCs. Many inflammatory genes are up regulated in chronic EAE-MSCs compared to peak EAE-MSCs. (D) Venn diagram illustrating similarities in DEGs found between naïve, peak, and chronic EAE-MSCs. (E) Top 10 transcriptional regulators (identified through MetaCore) associated with the DEGs found between peak EAE-MSCs and naïve MSCs (left) or chronic EAE-MSCs and naïve MSCs (right). Transcription factors (TF) are ranked according to their respective z-score (the level of connectivity of the TF to the DEG list), with the number of DEGs associated with that TF indicated above each bar.
|
Many of the DEGs found between peak and chronic EAE-MSCs were immune or inflammatory genes. These include some of the same chemokine and cytokine genes previously found elevated in EAE-MSCs in comparison to naïve MSCs. For example, chronic EAE-MSCs had elevated levels of expression of pro-inflammatory cytokine genes such as Cxcl1, Cxcl5, and Cx3cl1 compared to peak EAE-MSCs (Fig. 5C). Not all gene expression was elevated in chronic EAE-MScs. Several Toll-like receptor genes and compliment component genes were down regulated in chronic EAE-MSCs compared to naïve MSCs (Fig. 5C).
Overall, there was considerable consistency in the genes whose expression changed in BM-MSCs during EAE. For example, of the 2,337 DEGs identified between peak EAE-MSCs and naïve MSCs, 65% of these (1,498/2,337) were also differentially expressed between chronic EAE-MSCs and naïve MSCs (Fig. 5D). Furthermore, of the 5,837 DEGS found between chronic EAE-MSCs and naïve MSCs, 80% of these genes (2,979/3,736) were differentially expressed when comparing peak EAE-MSCs to chronic EAE-MSCs. In total, there were 988 common DEGs in BM-MSCs throughout all time points analyzed during EAE (Fig. 5D).
Bioinformatic analysis revealed a high degree of conservation in the transcriptional regulators associated with the DEGs identified in EAE-MSCs. For example, the transcription factor SP1 was most significantly associated with the DEGs identified in both peak EAE and chronic EAE-MSCs, and both peak EAE-MSCs and chronic EAE-MSCs showed a high degree of similarity in other transcription factors linked to their respective DEGs (Fig. 5E). Overall the current bioinformatic analysis suggests the changes in gene expression by BM-MSCs during EAE and the transcriptional regulators associated with these changes are highly conserved.
Go to :
Discussion
Utilizing RNA-Sequencing, genome-wide expression profiles of BM-MSCs derived from healthy and EAE mice have been analyzed and compared for the first time to provide a better understanding of the transcriptional and functional changes BM-MSCs undergo during inflammatory disease. Widespread gene expression changes were detected by RNA-Seq analysis in BM-MSCs derived from animals with EAE compared to naive cells derived from non-diseased animals. The degree of altered gene expression correlated with disease progression. Many of the most pronounced increases in gene expression were in genes involved in the regulation of the immune system and inflammatory processes. Bioinformatic analysis also identified important changes in EAE-MSCs in genes associated with pathways that influence neural cell fate and antagonize oligodendrocyte differentiation. Consistent with these changes in gene expression, conditioned medium from EAE BM-MSCs reduced the number of mature oligodendrocytes that developed in cultures of neonatal spinal cord, while conditioned medium from naïve BM-MSCs increased the number of oligodendrocytes in parallel cultures.
Mesenchymal stem cells have many characteristics that make them suitable for clinical application in a number of diseases (27). In order to ensure maximal benefit of such trials it is important that the most appropriate cells are used and the cells should be defined as effectively as possibly. The gene expression changes identified between naive BM-MSCs and MSCs derived from an animal model of multiple sclerosis (EAE) are potentially important given the majority of clinical trials for MS patients use autologous MSCs that are likely more equivalent to EAE derived MSCs. While preliminary results from these clinical trials report good safety data in transplanting autologous MSCs into MS patients, most report limited therapeutic efficacy (28–31) prompting questions about whether autologous MSCs from MS patients (MS-MSCs) are equivalent to allogeneic MSCs from healthy patients (naïve MSCs) in their functional characteristics and therapeutic potential (32).
Comparisons of the biological and functional characteristics of naïve and MS- MSCs have failed to reach consensus as to whether MS-MSCs show differences in characteristics like proliferation or cytokines secretion compared to naïve MSCs (33–35). One recent study reported MS-MSCs have less immunosuppressive functionality compared to naïve MSCs in-vitro, and utilized microarrays to profile and compare gene expression in MS-MSCs and naïve MSCs (36). Although there was a significant up-regulation of inflammatory genes in MS-MSCs, consistent with the RNA-Seq analysis of EAE-MSCs in the current study, the magnitude of changes was more limited. For example, less than 700 DEGs were seen in the MS study, compared to the greater than 2,000 DEGs seen in EAE (36). Interpreting genomic or functional data that compares MS-MSCs to naïve MSCs can be complicated as there are a range of variable that may alter the data and are difficult to control for. Factors such as genetic background or donor age may influence the functionality of BM-MSCs (37). Likewise, current or previous medication regimes, disease duration, and disease severity may affect gene expression levels in autologous BM-MSCs. Together such variables may make defining significant functional or gene expression changes in BM-MSCs derived from patients with diseases such as MS difficult. The utilization of a mouse model of disease such as EAE eliminates many of these variables, including genetic traits and prior disease history, and can provide clearer insight into the effects of disease on BM-MSCs.
A recent study comparing BM-MSCs derived from MOG EAE mice to naïve MSCs from healthy, age-matched mice demonstrated that the EAE-MSCs had a reduced ability to ameliorate functional deficits caused by EAE (18). This lack of therapeutic efficacy of EAE-MSCs was correlated with increases in the relative levels of pro-inflammatory cytokines and decreases in the levels of anti-inflammatory growth factors EAE-MSCs secreted relative to naïve MSCs. This cytokine expression data is consistent with the present RNA-Seq analysis showing EAE-MSCs up-regulate a wide variety of inflammatory genes associated with immune response and immune cell activation. The up regulation of gene expression for Cxcl1, Cxcl5, and IL-6 correlates directly with the increased expression of these cytokines in conditioned medium from EAE-MSCs (18).
The duration of disease appears to directly influence BM-MSC gene expression. For example, the number of DEGs was significantly increased in chronic EAE-MSCs versus naïve MSCs, and the magnitude of the gene expression changes tended to be higher in chronic EAE-MSCs compared to naïve MSCs. Increases in gene expression were particularly evident with inflammatory genes. These results are consistent with previous findings that BM-MSCs isolated from MOG-induced EAE mice during the chronic phase of the disease express elevated levels of inflammatory cytokines and have less immunosuppressive potential than BM-MSCs isolated from EAE mice during the peak of the disease (18).
In addition to modulating the immune system, MSCs have been suggested to influence neural cell fate. Several studies demonstrate that naïve MSCs have the capacity to enhance oligodendrocyte differentiation and maturation in-vitro and remyelination in-vivo in demyelinating animal models (8–10). It is currently unclear whether MSCs alter the fate of multipotent neural progenitor cells or directly promote the differentiation of oligodendrocyte precursors when transplanted in-vivo. Furthermore, the relative contributions of MSC mediated recovery in EAE from their capacity to influence neural cell fate and remyelination versus the ability of MSCs to suppress inflammation and the immune response is currently unclear (38). RNA-Seq analysis revealed a significant up regulation in gene expression of a number of TGFβ family members including BMPs in EAE-MSCs. Developmental studies have defined a central role for BMP signaling in inhibiting the development of mature oligodendrocytes from their precursors (39), and here we show that conditioned medium from EAE-MSCs no longer promotes the appearance of mature oligodendrocytes. These observations raise the possibility that the reduced therapeutic functionality of EAE-MSCs may be due in part to the diminished ability of these cells to promote oligodendrocyte maturation and subsequent remyelination. Future studies are required to better address how EAE alters the capacity of BM-MSCs to modulate neural cells and promote myelin repair, and to determine whether the loss of therapeutic functionality in EAE-MSCs stems from changes in their immunosuppressive versus neuroprotective potential. Exploring how EAE affects the ability of MSCs to modulate other CNS cell types, such as neurons and microglia, will also be important to further understand how disease may diminish the therapeutic capacity of MSCs.
Until there is a better understanding of how MS impacts the therapeutic potential of BM-MSCs, careful consideration should be given in using allogenic versus autologous MSCs in clinical applications to treat MS. Future studies exploring ex-vivo manipulations that could enhance or restore the therapeutic functionality of autologous MS-MSCs such as differential culture conditions or modification of gene expression may provide a more acceptable approach to an effective therapy. The present RNA-Seq analysis of EAE MSCs provides new pathways and potential targets that could be exploited to achieve this effect.
Go to :
Conclusions
RNA-Seq analysis of BM-MSCs in EAE helps to support newly emerging evidence that disease alters the functional characteristics and therapeutic capacity of MSCs. EAE-MSCs show clear changes in gene expression patterns relative to naïve MSCs, including up-regulation of inflammatory genes. In addition, EAE-MSCs differentially modulate neural cell development and fail to promote the formation of mature oligodendrocytes in-vitro. We show that inflammatory disease dramatically alters BM-MSCs at both a transcriptional and functional level, raising concern about the efficacy of using autologous MSCs to treat MS.
Go to :
 XML Download
XML Download