

 PDF
PDF Citation
Citation Print
Print


Introduction
Mesenchymal stem cells (MSCs) are multipotent and differentiate into a variety of specialized cells. Although they are less potent in differentiation than embryonic stem cells, MSCs are far easier to obtain with much less concern regarding ethical issues and are therefore useful resources for cell therapies and regenerative medicine. By December 2017, near seven thousand clinical trials had been registered or completed worldwide (1). MSC infusion appears to be tolerated without severe adverse effects, raising expectations that their application might be expanded to a wider variety of aging-associated dysfunctions.
In most clinical trials using MSCs, approximately 108 cells in total or 1×106 cells/kg recipient weight are administered multiple times in a therapy (2). This large number of cells is obtained through more than 25 doublings from a single cell, which represents a large portion of the replicative potential of MSCs. Stem cells are generally characterized as able to multiply for long periods without changes in their pluripotency. However, MSCs lose functionality during in vitro cultivation and during aging (3). Like other normal human cells such as fibroblasts, MSCs are subject to limitations in doubling capacity (or replicative life span) and enter a stage called replicative senescence, which is mainly attributed to telomere shortening (4). In addition, late-passage human MSCs tend to lose their stemness property, especially for multi-lineage differentiation (5–7). As a result, transplantation of late passage cells would result in null or even undesirable outcomes in the recipient. After long-term passage, MSCs lose their adipogenic, osteogenic, and neurogenic differentiation potential and may also change lineage upon the same differentiation cue (6, 8). Late-passage MSCs also tend to express lower levels of trophic factors when transplanted into animal models, reducing the efficacy of cell therapy (9). These characteristics clearly impose an unignorable limitation to stem cell therapies in aged individuals who have a greater need for them. In many studies, MSCs from aged human donors have exhibited reduced growth rate and capacity, as well as decreased potential for differentiation (7, 9, 10). There is therefore an urgent need for methods to decelerate replicative senescence of MSCs.
The main cause of the shortened replicative life span of normal cells is accumulation of reactive oxygen species (ROS). ROS-induced damage to DNA and mitochondria is especially problematic. Damage to DNA accelerates telomere shortening (11), whereas damage to mitochondria escalates a vicious cycle of ROS generation and further mitochondrial damage. Damaged electron transport chains leak a large amount of electrons, leading to a high level of ROS, which in turn cause further damage (12). Therefore, mitochondria are conceivably both victims and culprits in shortening of the replicative life span and acceleration of cellular senescence. Importantly, mitochondrial functionality and ROS also affect differentiation. Osteogenesis induction accompanies an increase in the efficiency of mitochondrial respiration and a decrease in ROS level through an increase in mitochondrial DNA copy number and the amount of respiratory and antioxidant proteins (13). Adipogenesis has also been shown to accompany mitochondrial biogenesis and oxygen consumption, although it requires an increase rather than decrease in ROS (14). A high level of ROS also appears to be necessary for chondrogenesis, at least in mice MSCs (15). Although it is not clear whether these changes are consequences or triggers of osteogenesis and adipogenesis, these findings indicate that mitochondrial function and ROS level are strongly associated with differentiation potential and lineage determination of MSCs. On a practical level, these findings triggered tests of the impact of antioxidants on stem cells used in therapies, although no single antioxidant by itself has been shown to be sufficiently effective. However, some chemicals showed promise and most of these appeared to work through substantial elevation of endogenous anti-oxidative capacity (3, 16).
Nicotinamide (NAM), an amide form of vitamin B3, is a potent inhibitor of sirtuins, a family of NAD+-dependent deacetylases. The sirtuin family proteins, of which SIRT1 is the best known, play critical roles in cellular and organismal health and longevity (17), and NAM has been predicted to negatively affect cell viability. However, in cells NAM is rapidly converted to NAD+ through a salvage pathway, and therefore treatment with NAM has been found to increase SIRT1 activity (18). As a result, NAM appears to protect cells against oxidative stress in a number of studies, including ours. For example, administration of 5 mM NAM caused a decrease in mitochondrial superoxide levels and resulted in substantial extension of proliferative potential in normal fibroblasts (19). The treated cells showed a decrease in mitochondria content but an increase in their quality as evidenced by lower levels of ROS generation and oxidative damage to mitochondrial proteins. These changes might be due at least in part to activation of autophagy (and mitophagy) through SIRT1 mobilization driven by the NAM-induced increases in the NAD+/NADH ratio (20).We reasoned that this property of NAM could be used as a strategy to enhance mitochondrial quality and expand the doubling capacity of MSCs, and therefore tested NAM in human bone marrow stem cells (BMSCs). Our results showed that NAM treatment indeed increases the replicative life span of human BMSCs. Furthermore, NAM treatment increased the differentiation potential of osteogenesis, adipogenesis, and chondrogenesis, and suppressed the adipogenic switch in the tested BMSCs. These results indicate that NAM might be an important biological agent to broaden the use of MSCs for cell therapies.
Materials and Methods
Isolation and expansion of human BMSCs
MSCs were isolated from the bone marrow of three different healthy individuals aged 31, 39, and 50 years, respectively (IRB#: KPH 2009-01). Briefly, bone marrow samples were layered on Ficoll-Paque solution (GE Healthcare, Buckinghamshire, UK) at a ratio of 1:1 and centrifuged at 2,200 rpm for 20 min. The interface layer was collected, washed twice in phosphate-buffered saline (PBS), re-suspended in MSC growth medium (MSCGMTM) (Lonza, Alpharetta, GA, USA), plated in 100-mm culture dishes, and incubated in a humidified atmosphere of 5% CO2 at 37°C. The medium was changed after 48 h and floating cells were removed. Cells were sub-cultured in MSCGMTM containing 10% fetal bovine serum (FBS) (Lonza), 1% antibiotic-antimycotic (ThermoFisher Scientific, Waltham, MA, USA), and 1% glutamine. Bone marrow MSCs were identified by positivity for the surface markers CD45, CD34, CD29, CD73, CD105, CD44, and CD90 by flow cytometry using FACSCalibur (BD Biosciences, San Jose, CA, USA) and CellQuest software (BD).
Population doubling and nicotinamide treatment
The three BMSC lines had different passage histories before this study. However, at the point of cultivation for this study counting of the population doublings (PD) started anew as PD 0. Subcultures were made every 6~7 days at a dilution of 1:4 during early passages and 1:2 during late passages. The number of PDs (n) was calculated using the equation n=log2(F/I), where F and I are the numbers of cells at the end and at the beginning of the passage (usually 2.0×105), respectively. Cell numbers were counted using an AccuChip kit (Science Services, Munchen, Germany) and the growth curve was plotted using Sigmaplot software (Systat Software Inc., Richmond, CA, USA). Nicotinamide (NAM) was added to the medium at a concentration of 5 mM and maintained in the medium throughout the culture. All chemicals, including NAM, were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated.
Osteogenic differentiation
BMSCs were seeded in 24-well plates at 100% confluency. After 2 days, the medium was changed to osteogenic induction medium (OIM) composed of 10 mM β-glycerol phosphate, 50 μM ascorbic acid, 0.01 μM dexamethasone, and 10 FBS in MSCGM. After incubation for 12 days the cells were washed with PBS and fixed with 4% paraformaldehyde, and osteogenic differentiation was determined by staining with Alizarin Red S (ARS) for 20 min to detect extracellular calcium deposits. For quantification of calcium deposits, cells stained with ARS were extracted in 10 % acetic acid for 30 min at room temperature and for 10 min at 85°C. After neutralization with ammonium hydroxide, 150 μl aliquots were transferred to 96 wells, and absorbance at 405 nm was determined in spectrometer.
Adipogenic differentiation
BMSCs were seeded in 24-well plates at 100% confluency and incubated in adipogenic induction medium (AIM; FBS-supplemented MSCBM containing 33 μM biotin, 17 μM pantothenate, 5 μg/ml insulin, 0,25 μM dexamethasone) for 3 days in the presence of 10 μM rosiglitazone and 0.5 M 3-isobutyl-1-methylxathine, and for a further 6 days or more in their absence. To determine adipogenesis, the cells were fixed with 4% paraformaldehyde and stained with 0.2% Oil red O in isopropanol for 2 h. Cells were washed with 60% isopropanol and water and observed under the microscope. For quantification of lipid accumulation, stained oil droplets were dissolved in isopropanol for 15 min and absorbance at 518 nm was determined using a spectrophotometer.
Chondrogenic differentiation
BMSCs were induced to chondrogenesis in a high cell density spheroid using a commercially available medium. Briefly, 2.5×105 cells were aliquoted and re-suspended in PRIME-XV® Chondrogenic Differentiation XSFM (Irvine Scientific, Santa Ana, CA, USA) in 15 ml conical tubes. The tubes were centrifuged and placed in a CO2 incubator for 24 h, after which the aggregates were gently poured with the medium to allow them to float freely. The tubes were kept in the incubator for 15 days with a change of PRIME-XV® Chondrogenic Differentiation XSFM in every 2~3 days. The expression of cartilage molecules in chondrogenic spheroids was assayed by fixing with 4% formalin and staining with 1% Alician Blue solution prepared in 0.1 N HCl for 30 minutes. The spheroids were visualized under light microscopy and photographed.
Measurement of mitochondrial content and ROS
To quantify mitochondrial content, cells were stained with 50 nM nonyl acridine orange or 100 nM MitoTracker Red (both from ThermoFisher Scientific) for 30 min at 37°C in the dark and then subjected to flow cytometry with 488 nm excitation and 585 nm or 530 nm emission. To quantify ROS levels, cells were incubated with either 5 μM dihydroethidium (DHE), 0.1 μM MitoSox (Thermo-Fisher Scientific), or 15 μM dihydrorhodamine (DHR) 123 for 30 min, washed with PBS, collected in PBS containing 1 mM EDTA, and analyzed using FACSCalibur (BD Biosciences). Values of mean fluorescence intensity were plotted using SigmaPlot 9.01 software.
Measurement of cellular free NAD+/NADH ratio
Cellular NAD+/NADH ratio was determined by lactate/pyruvate oxidase assays. Briefly, 10 μl of the culture medium or lactate standard solution was mixed with 196 μl of the assay buffer (0.1 M citrate, 1 mg/ml BSA, 0.1% CaCl2, 0.02% NaN3, adjusted to pH 6.5 with 1 M Na2HPO4), 1 μl of 2 mU/μl lactate oxidase stock (lactate oxidase enzyme dilution buffer [10 mM KH2PO4, 10 μM FAD, adjusted to pH 7.0 with KOH], 1 μl of 0.5 U/μl peroxidase solution), and 2 μl of 5 mM Amplex UltraRed stock (ThermoFisher Scientific). Assay mixtures were incubated for 30 min at 37°C and fluorescence was read at excitation/emission of 535/590 nm. For the pyruvate oxidase assay, 10 μl of the culture medium or pyruvate standard solution was mixed with 196 μl of the assay buffer (50 mM KH2PO4, 1 mg/ml BSA, 0.2 mM thiamine pyrophosphate, 10 μM FAD, 0.97 mM EDTA, 9.8 mM MgCl2, 0.02% NaN3, adjusted to pH 6.5 with 1 M NaOH), 1 μl of 2 mU/μl pyruvate oxidase solution, 1 μl of 0.5 U/μl peroxidase stock, and 2 μl of 5 mM Amplex UltraRed stock. The assay mixtures were incubated for 30 min at 37°C and fluorescence was read at 535 nm (excitation) and 590 nm (emission). For the calculation of the ratio of free NAD+/NADH, the following equation was used: free NAD+/NADH=[pyruvate]/[lactate]×1/K, K=1.11×10−4.
In situ staining of senescence-associated β-galactosidase activity
Cells were fixed with 3% formaldehyde and washed in PBS (pH 6.0) containing 2 mM MgCl2. After incubation overnight at 37°C in β-galactosidase staining solution (1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside, 5 mM K3Fe[CN]6, 5 mM K4Fe[CN]6, and 2 mM MgCl2), cells were examined using bright-field microscopy.
Western blot analysis
Cells were lysed with RIPA buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with NaF, NaVO4, and a protease inhibitor mixture. Typically, 30~40 μg of total protein was separated by SDS-PAGE, transferred to nitrocellulose membranes, and blotted with antibodies against human acetylated p53 (Cell Signaling Technology, Beverly, MA), p53, SIRT1, LC3, β-actin, or ERK1/2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein bands were visualized using horseradish peroxidase-conjugated secondary antibodies and SuperSignal West Femto substrate (ThermoFisher Scientific).
qPCR for mRNA quantification
Total RNA was isolated using RNeasy® Mini kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. A total of 5 μg of RNA was converted to cDNA using MuLV reverse transcriptase (BEAMS bio, Sungnam, South Korea) and oligo (dT) primer (Bionics, Daejeon, South Korea), and 1/40 volume of the cDNA reaction was used as the template for PCR in MyiQTM (Bio-Rad, Hercules, CA, USA) using primers for human cDNA sequences of RUNX2, SOX9, COL1A1, COL2A1, and β -actin. PCR results were quantitatively analyzed by iQ5 software. Sequences of the primers used will be provided upon request.
Statistical analysis
Most quantifications were performed with two independent measurements of samples from two different experiments, and mean±S.E.M values were presented. Intergroup comparison of the mean values was performed by one-way analysis of ANOVA using InStat 3.06 (GraphPad Software Inc., San Diego, CA, USA). Significant differences are indicated as *(p<0.1) and **(p<0.01).
Results
Enhancement of proliferation capacity of BMSCs by NAM treatment
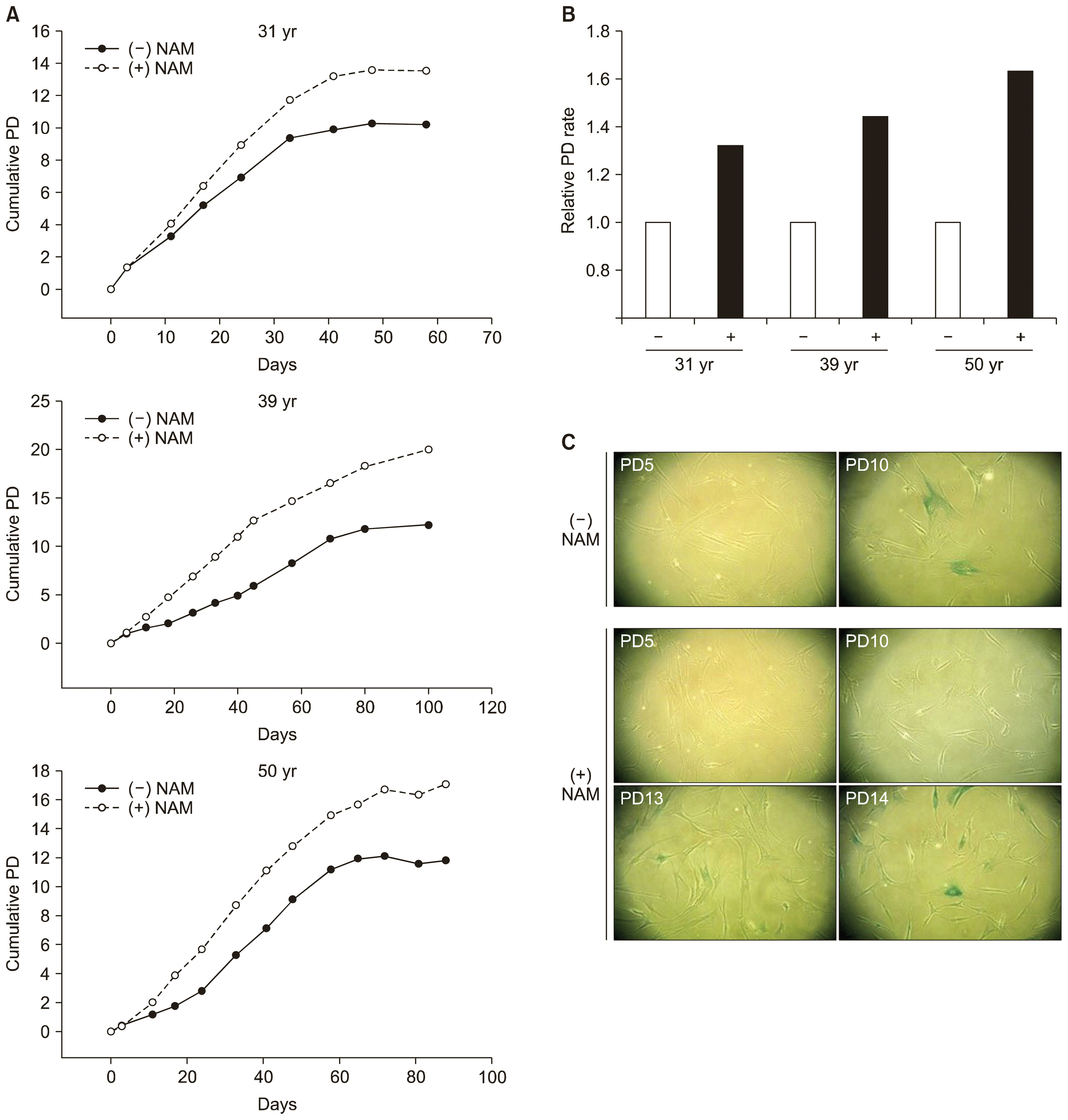
NAM treatment was shown to extend the replicative life span of human fibroblasts (19). We therefore examined whether NAM exerts a similar positive effect on the proliferation of human BMSCs. Three different lines of BMSCs from donors aged 31, 39, and 50 years respectively were cultivated in MSCGM in the presence of 5 mM NAM from PD 0. The three lines showed different rates of proliferation without close correlation with donor age. Importantly, replicative life span of all the lines was positively affected by NAM treatment (Fig. 1A) and was extended by 33~70%, which is similar to results achieved in NAM-treated fibroblasts. Furthermore, the proliferation rate (population doubling rate) increased in all the lines in the presence of NAM as shown by the steeper increase of the growth curve (Fig. 1A) and higher PD rate (Fig. 1B). This effect was not observed in the study on fibroblasts (19). Quantification of cell division using CFSE, a fluorescent dye that becomes membrane impermeant once it enters the cell, showed that NAM-treated cells underwent two–three times more divisions during 10 doublings of the cells incubated in the absence of NAM (Supplemental Fig. 1A). In addition, cell viability also increased (Supplemental Fig. 1B), demonstrating that NAM protected BMSCs. The BMSCs appeared to be less stable than fibroblasts during the in vitro cultivation, showing only 82% viability in 3-day culture. This value increased to 94% in the presence of NAM. Therefore, the higher proliferation rate is attributed to both faster cell division and enhanced cell viability. Moreover, the expansion of proliferation potential was apparently linked to a delay in the onset of replicative senescence. The expression of SA β-Gal, a frequently used marker of senescence (21), was attenuated until near the end of the extended life span (Fig. 1C; compare PD10 of (−) NAM panel and PD13 of (+) NAM panel). Above all, these results indicate that NAM postpones the onset of senescence of BMSC populations and drives them to expand far beyond the designated replicative life span. For most of the studies, all three BMSC lines were tested but the results for the 31-year-old donor line are presented in Fig. 1A as representative findings.
Effect of NAM on the levels of ROS and mitochondria in BMSCs
The level of ROS is an important determinant of replicative life span and viability of primary cells. In this regard, mitochondria play a critical role because they are a major source of ROS and dysfunctional mitochondria produce more ROS (12). In addition, mitochondrial function deteriorates during long-term MSC cultivation (22). We examined whether ROS level and mitochondrial content of BMSCs are affected by NAM treatment. NAM treatment appeared to suppress ROS in BMSCs. Levels of mitochondrial superoxides (MitoSox), hydroxyl radical (DHR123), and cytosolic superoxides (DHE) increased by 30~90% during 10-day cultivation (Fig. 2A). However, this increase was significantly lower or even suppressed in cells cultivated in the presence of NAM (Fig. 2A). We previously reported that treatment of human fibroblasts with 5 mM NAM increased the cellular level of NAD+ and the ratio of NAD+/NADH in the cytosol as well as in mitochondria, and accordingly decreased electron transport through the mitochondrial respiratory chain and reduced ROS generation from mitochondria (23). The increase in the cellular NAD+/NADH ratio also increased the activity of SIRT1, which in turn activated mitophagy (20). In BMSCs, the NAD+/NADH ratio increased continuously during 9 days of NAM treatment (Fig. 2B). Furthermore, activation of SIRT1 and autophagy also occurred in BMSCs treated with NAM as evidenced by the decreased p53 acetylation level and increased formation of LC3 type II (which is induced by SRT1720, a SIRT1 activator) (Fig. 2C). During the 10 days of NAM treatment, mitochondria content decreased by nearly 25% suggesting increased mitophagy (Fig. 2D). Together, these data demonstrate that NAM treatment reduces ROS generation in BMSCs, most likely through modulation of mitochondria activity and content analogous to the response in fibroblasts.
Effect of NAM on osteogenesis of BMSCs
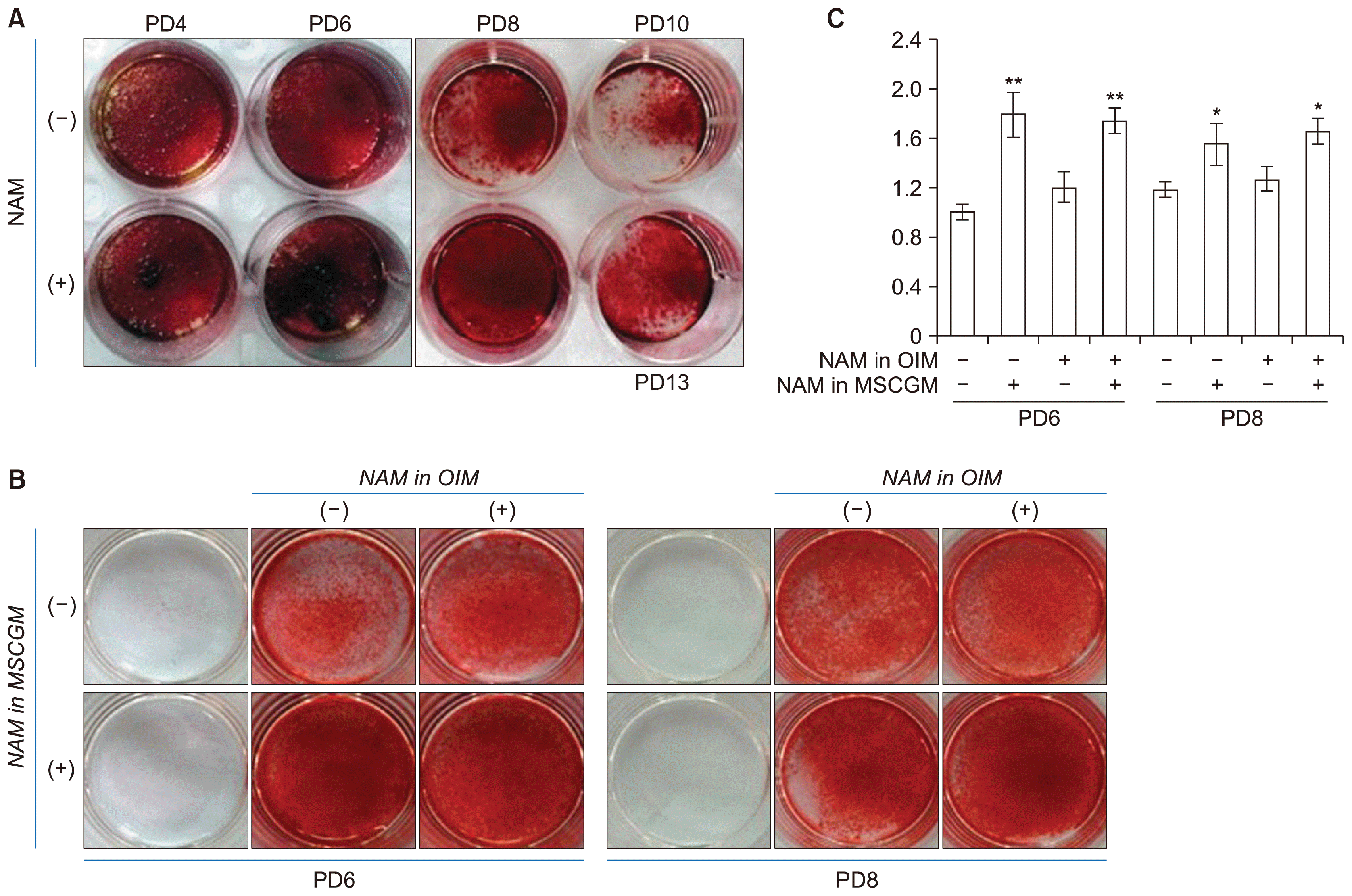
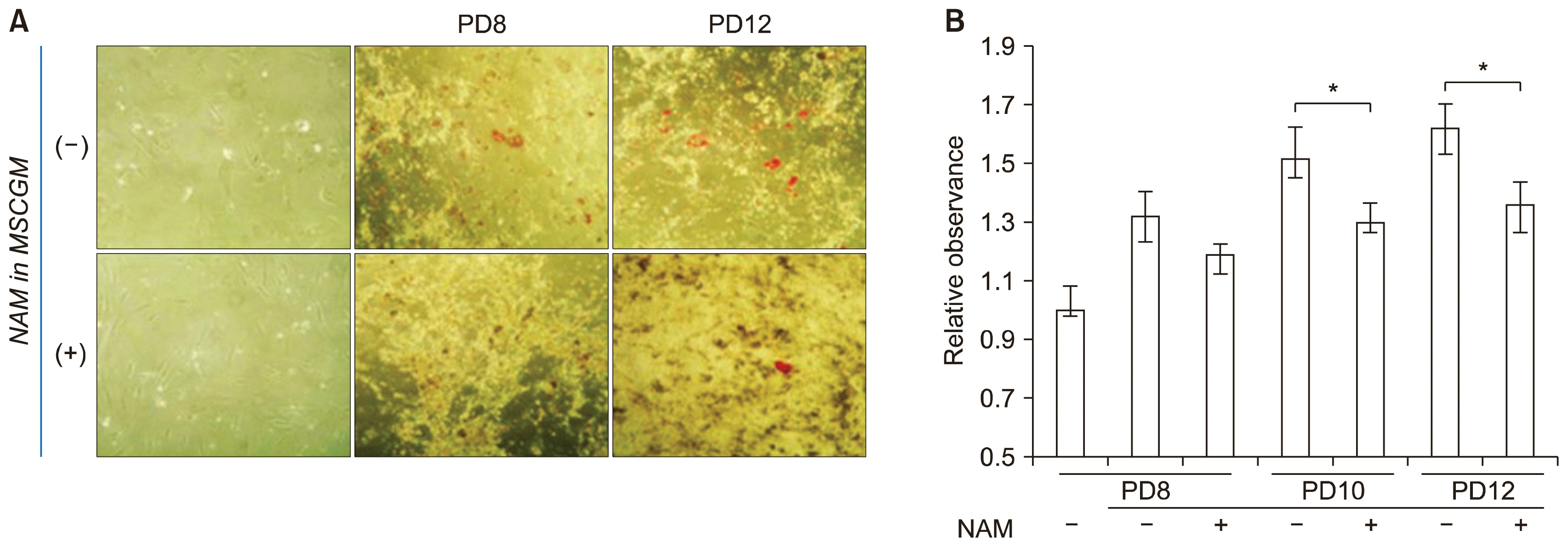
We also investigated whether NAM treatment affects BMSC differentiation. First, cells cultured in the presence or absence of 5 mM NAM for different number of passages were induced to undergo osteogenesis and their calcium deposition was compared. Cells cultured in the absence of NAM proliferated to slightly over PD 10 whereas cells cultured in the presence of NAM were passaged until PD14, as shown in Fig. 1A. In both conditions osteogenesis appeared to peak at PD 6 and declined in later passages, but higher levels of calcium deposition were obvious in cells cultured in the presence of NAM (Fig. 3A, lower wells). Furthermore, osteogenesis was sustained throughout their extended life span (PD13). These results indicate that NAM treatment not only extends the life span of the tested BMSCs but also enhances and sustains their osteogenic potential.
In this experiment, NAM was present during both the continued cultivation and differentiation. Therefore, it is not clear whether NAM facilitated better sustenance of osteogenesis or induced osteogenesis to a higher level during the differentiation process. To distinguish between these alternatives, NAM was added to either the growth medium only or the osteogenic induction medium only and the level of osteogenesis was compared. Cultures maintained in the absence or presence of NAM (for PD6 or PD8) were divided into two groups and each was induced to undergo osteogenesis in the absence or presence of NAM. The presence of NAM during the induction period had little effect on the level of calcium deposit (compare (−) and (+) in NAM in OIM, Fig. 3B and 3C), whereas the presence of NAM during cultivation caused enhanced calcium deposition (compare (−) and (+) in NAM in MSCGM, Fig. 3B and 3C). The same results were obtained for early passage (PD6) and late passage (PD8) cells. These results indicate that NAM treatment might help cells maintain their osteogenic potential during cultivation.
Effect of NAM on adipogenesis and chondrogenesis of BMSCs
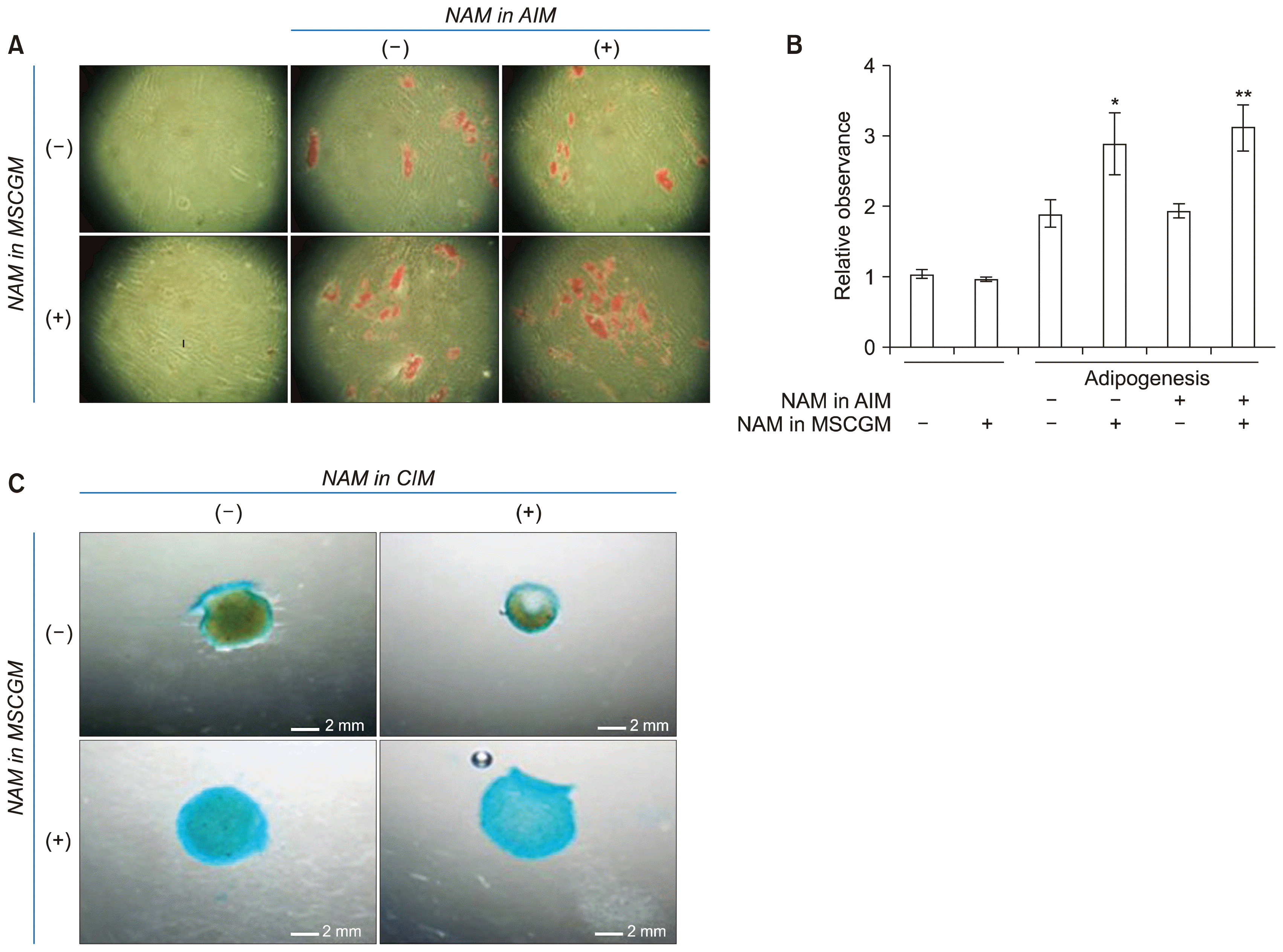
The results for osteogenesis suggest that NAM might attenuate the passage-induced decline of differentiation capacity of BMSCs. To determine whether other types of differentiation are similarly affected the same line of BMSCs used in the osteogenesis experiments was induced to either adipogenesis or chondrogenesis. As shown in Fig. 4, differentiation of both lineage types was positively affected by the presence of NAM during cultivation. Again, the presence of NAM during the differentiation processes showed no effect. This was apparent when the red precipitates of lipid droplets in the panels of (−) and (+) in NAM in AIM and in NAM in MSCGM were compared (Fig. 4A). Quantitative comparison of the levels of adipogenesis also indicated that NAM was effective only when present in the growth medium (Fig. 4B). The darker blue color of the cartilage spheroids formed in NAM (+) MSCGM in Fig. 4C (regardless of the presence of NAM in differentiation) also indicates the effect of NAM on chondrogenic differentiation. Overall, the enhancement and sustenance commonly seen in the three different types of differentiation indicate that NAM treatment helps BMSCs sustain their stemness potential during continued cultivation, and thereby allows expansion of the differentiation potential of BMSCs in parallel to the increase in proliferation potential.
Effects of NAM on the level of differentiation factors during osteogenic and chondrogenic induction
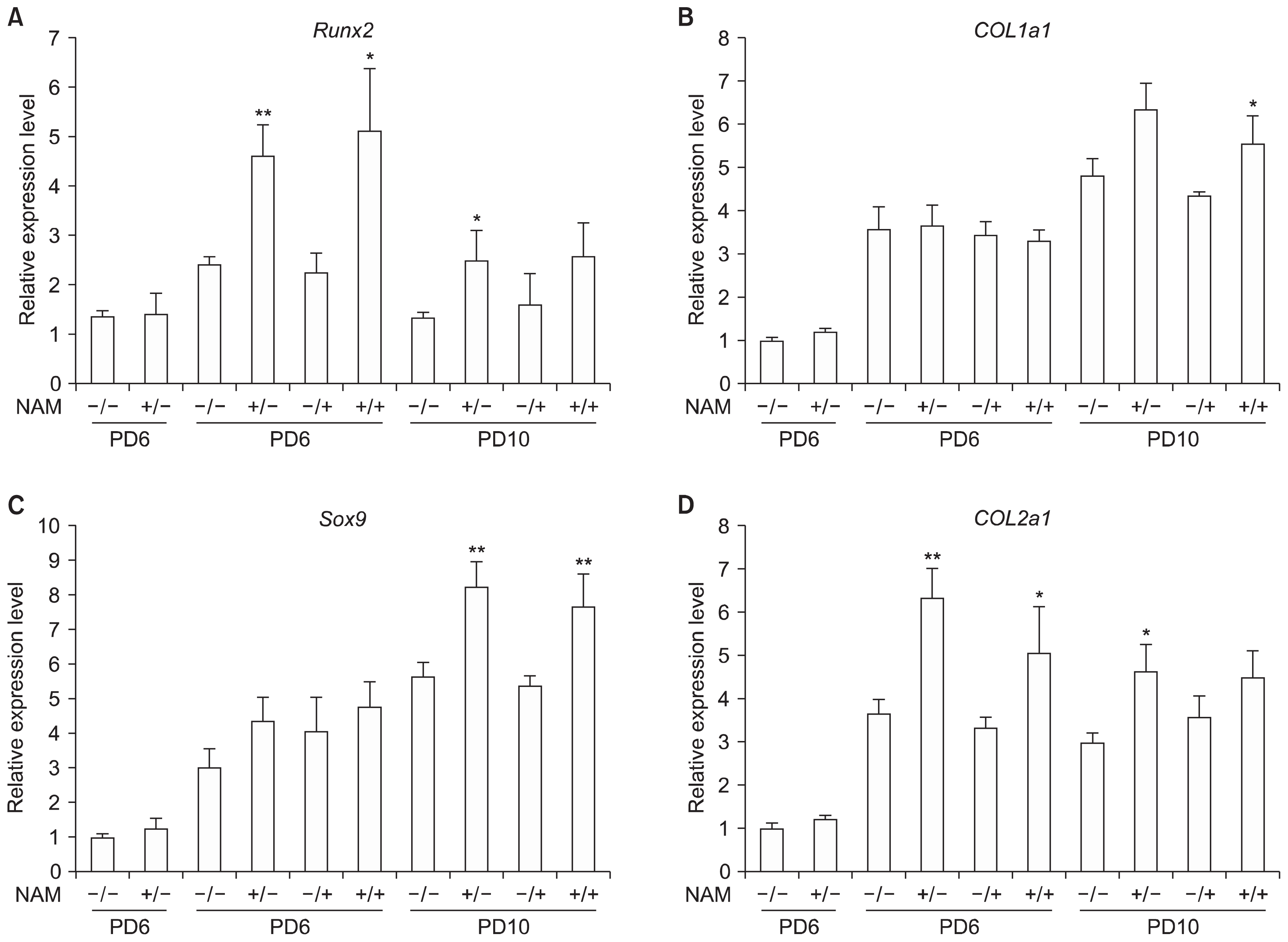
Differentiation processes in cells cultured in the presence of NAM were examined by quantitative analysis of the levels of key transcription factors and markers of osteogenesis and chondrogenesis. Runt-related transcription factor (Runx) 2 is a key osteogenic transcription factor that plays a determinant role in early osteogenic differentiation (24), and collagen type I alpha1 chain (COL1a1) is one of its targets (25). Sry-related transcription factor (Sox) 9 is a key chondrogenic transcription factor (26), while COL2a1, one of its targets, is an early and abundant marker of chondrogenic differentiation (26). Cells at PD 6 or PD 10 were induced to either osteogenesis or chondrogenesis and collected at day 7 (for osteogenesis) or day 3 (for chondrogenesis) of differentiation for analysis of the mRNA levels of Runx2, COL1a1, Sox9, and COL2a1 by real time-PCR. As expected, in most cases expression of these factors was induced at higher levels in cells cultivated in the presence of NAM but not in cells differentiated in the presence of NAM (Fig. 5). Moreover, the expression of these factors was sustained (for Runx2 and COL2a1) or even increased (for Sox9 and Col1a1) in the later passages. These results provide molecular evidence supporting the differentiation enhancing and sustaining effects of NAM.
Effect of NAM on adipogenic switch of BMSCs
It is generally known that excess ROS impair osteogenic differentiation (27). In addition, MSCs from aged donors experience a shift in differentiation lineage from osteogenesis to adipogenesis (adipogenic switch) during in vitro passage (22). We determined whether the extension of replicative life span by NAM treatment affects the adipogenic shift using a BMSC line from the oldest of our donors (Fig. 1C). BMSCs cultivated in the absence or presence of NAM were induced to osteogenesis and assayed for adipogenesis by Oil-Red-O staining. In both populations, lipid droplets were formed upon osteogenic induction and became more apparent in later passages (PD 12) (Fig. 6A and 6B). However, lipid formation was lower in the population cultured in the presence of NAM, indicating that the passage-dependent adipogenic shift is attenuated by NAM treatment.
Discussion
Our results demonstrated that treatment with 5 mM NAM during in vitro cultivation of human BMSCs increases their replicative life span as well as their differentiation potential in osteogenesis, adipogenesis, and chondrogenesis. NAM reduced the levels of ROS and mitochondria during BMSC proliferation, and this effect may underlie the delayed imposition of replicative senescence of BMSCs. A lower level of ROS generation from mitochondria has been observed in NAM-treated fibroblasts, and was proposed to be responsible for reduced telomere shortening and the delayed onset of replicative senescence (19). Therefore, NAM treatment is certainly beneficial for maintaining pluripotency of BMSCs. Moreover, since the changes in ROS level and most of the altered mitochondrial functions in NAM-treated cells are induced by a high level of NAD+ or NAD+/NADH ratio, which drives sirtuin activation and also attenuates ROS generation from the respiratory chain (23), NAD+-precursor molecules such as nicotinic acid, nicotinamide mononucleotide, and nicotinamide riboside are expected to have similar positive effects on BMSC stemness potency.
As emphasized above, the NAM-induced extension of replicative life span can easily be primarily attributed to the decrease in ROS. However, there are multiple possible mechanisms for the increased and sustained differentiation potency of BMSCs. First, this might also result from a reduced ROS level. The differentiation potential of MSCs decreases with age and this is likely associated with increased ROS levels in aged tissues (7). ROS inhibit osteogenic differentiation, and oxidative stress must be decreased for osteogenic differentiation to proceed (13). Therefore, the NAM-mediated decrease in ROS likely increased osteogenesis. However, this does not easily explain the increase in adipogenesis. ROS generated by the mitochondrial respiratory chain were shown to be essential for the activation of adipogenic transcription factors and adipogenesis (28). Furthermore, inhibition of mitochon drial respiration suppresses adipogenesis (14). These findings suggest that NAM, which suppresses mitochondrial ROS generation, would generate a cellular condition that favors osteogenic differentiation but discourages adipogenesis. And, our previous studies rule out the possibility of the causative effect of the reduction in cellular ROS level alone. The treatment of n-acetyl cysteine (NAC) could induce a decrease in cellular ROS level in tested human fibroblasts. However, this was not accompanied by an extension of replicative life span or a delay in the onset of senescence (19). Another, and more plausible, contributor to the differentiation potentiation may be the NAM-mediated extension of life span itself. The potentiation, i.e., increase and sustenance, of differentiation might simply be a phenomenon that results from the delay in cellular senescence. In this way, not only the expression of senescence phenotypes but also the decline of the differentiation program is postponed. This possibility is supported by the increased differentiation of all tested lineages, namely osteogenesis, adipogenesis, and chondrogenesis. The effectiveness of NAM present in the growth medium, but not in differentiation-induction medium, and the sustained expression of key marker genes of osteogenesis and chondrogenesis in late passage cells also support this possibility. However, considering the current inconsistency in reports on the levels of differentiation in different lineages during in vitro cultivation and aging (29, 30), the involvement of certain cellular factors and molecular programs may need to be considered.
A direct involvement of SIRT1 should also be considered. SIRT1 is known to play direct roles in both the proliferation and the differentiation of stem cells; its knockdown decreases both proliferation and differentiation of MSCs and overexpression has the opposite effect (31). SIRT1 activation contributes to the enhancement of mitochondrial quality by increasing mitochondrial turnover, i.e., biogenesis and mitophagy (32), and thereby attenuates the generation of ROS. Further, SIRT1 activation enhances MSC osteogenesis by directly modulating the activity of Runx2 through deacetylation (33), and downregulates MSC adipogenesis by directly inhibiting PPARγ, a key adipogenic transcription factor (34). SIRT1 level and activity is downregulated during both aging and senescence (18). This pro-osteogenic and anti-adipogenic effect of SIRT1 activity does not fit with the simultaneous potentiation of osteogenesis and adipogenesis but is consistent with the change in the adipogenic switch of differentiation of BMSCs induced by NAM. It is therefore possible that the decreased adipogenic switch in the cells cultured in NAM can be explained by NAM-mediated SIRT1 activation. Moreover, NAM treatment has also been shown to activate SIRT3, which deacetylates mitochondrial proteins (35). SIRT3-mediated deacetylation of mitochondrial proteins is associated with increased efficiency of the electron transport chain (36) and closure of the mitochondria permeability pore (mPTP), which enhances the level of ROS (37). Furthermore, SIRT3 activates superoxide dismutase 2 (SOD2) (38). By modulating the level of mitochondrial ROS, SIRT3 activation by NAM would certainly affect the potential of MSCs for replication and differentiation. However, a direct role of SIRT3 in MSC functionality has not been reported.
Repeated cell passage of MSCs to obtain a sufficient number of cells to achieve therapeutic effects generally decreases the quality of MSCs and reduces their therapeutic effectiveness. Chemical and physical means to increase the potential of MSC in cell therapies have been reported (3). These mostly involve a direct reduction in cellular ROS or oxidative stress or an increase in anti-oxidative capacity. Activators of autophagy and proteolysis are examples of the former. Hypoxia and hydrogen gas may also work by reducing oxidative stress. Activators of anti-oxidant enzymes such as Nrf2 are the examples of the latter (3). Although it is not a direct antioxidant or scavenger, NAM may work by allowing higher NAD+ redox and modulating mitochondria activity and quality, and might be an excellent additive in the cultivation of stem cells for cell therapies.

 XML Download
XML Download