

 PDF
PDF Citation
Citation Print
Print


Introduction
Recent progress in our understanding of brain development has significantly altered concepts for treating neurodegenerative diseases, including those that affect the retina to cause blindness. Contrary to previous thought, it is now recognized that neurons are generated in the sub ventricular zone (SVZ) of the lateral ventricle, and the sub-granular zone (SGZ) in the hippocampus from neural stem cells (NSC) of glial origin throughout life (1). Outside these discrete regions in the mammalian CNS, including the retina, active neurogenesis has not been reproducibly demonstrated under normal conditions (2). However, rare neurogenic changes are observed in the injured adult mammalian retina; the source of injury-induced neurogenesis is traced to Müller glia (MG) (3–5). Recent observations that mammalian MG possess NSC properties and are able to generate retinal neurons in vitro and upon transplantation in vivo posit these cells as the potential target for regenerating diseased or injured photoreceptors and retinal ganglion cells (6). While growing evidence confirms that MG possess evolutionarily conserved neurogenic potential, studies from various labs have observed that, unlike their lower vertebrate counterparts, the injury or disease-activated mammalian MG proliferate, but that they convert to neurons rather infrequently (4, 7, 8). Whether or not the converted neurons are functional, make synaptic connections, and survive for the long term remains unknown. The challenge that remains is how to unlock the neurogenic potentials of the mammalian MG in vivo. This may essentially involve approaches to reprogram these cells to dedifferentiate and regain their lost ability to generate neurons or trans-differentiate them into neurons.
Go to : 
Neurogenic potentials of MG in mammals
To address this challenge, the neurogenic potentials of MG need to be evaluated against the backdrop of two models, with the understanding that examples from lower vertebrates would constitute a framework, but that solutions will be unique to the mammalian retina: (1) SVZ/SGZ model, where MG have NSC properties and thus the capacity for neuronal differentiation like radial glia-derived NSCs in SVZ and SGZ. Given the observations that the adult NSCs are responsive to environmental cues for neuronal differentiation (1), MG in this model should be amenable to directed differentiation along neuronal lineage through the manipulation of niche-based signals by recombinant growth factors and/or small molecules. (2) Extra SVZ/SGZ model, where MG may have neurogenic potential like parenchymal astrocytes but incapable of neuronal differentiation, presumably under the influence of the non-neurogenic niche. For example, both MG and astrocytes express NSC regulators such as Sox2 (MG) and Pax6 (astrocytes) and when cultured in the presence of mitogens tend to differentiate along neuronal lineage (2, 6, 9, 10). However, in vivo, although they proliferate and express neural progenitor markers such as Nestin in response to injury, they maintain their glial phenotype, with rare expression of neuronal markers (2, 5, 10). Given their stable glial identity for supporting neurons despite expressing some of the NSC-specific genes, a trans-differentiation approach for the lineage conversion of MG has emerged as a practical option in this model. This notion is supported by recent observations that injury-activated resident astrocytes differentiate into functional neurons upon enforced expression of NSC regulator, Sox2 and/or proneural gene, Ascl1 (11). Regardless, information gained in deciphering the two models will be useful. In both cases, knowledge of the developmental mechanism for the lineage switch, the reaction of MG to different kinds of injuries, phenotypes of the activated MG in spatial and temporal contexts, transcriptional and epigenetic status, and niche-related signals will be critical. These approaches will reveal whether the roadblock to regeneration is cell-intrinsic or cell-extrinsic or both, information that will be essential in formulating strategies for making MG-dependent therapeutic regeneration practical and clinically applicable. Toward this effort, the following issues should be addressed (Fig. 1).
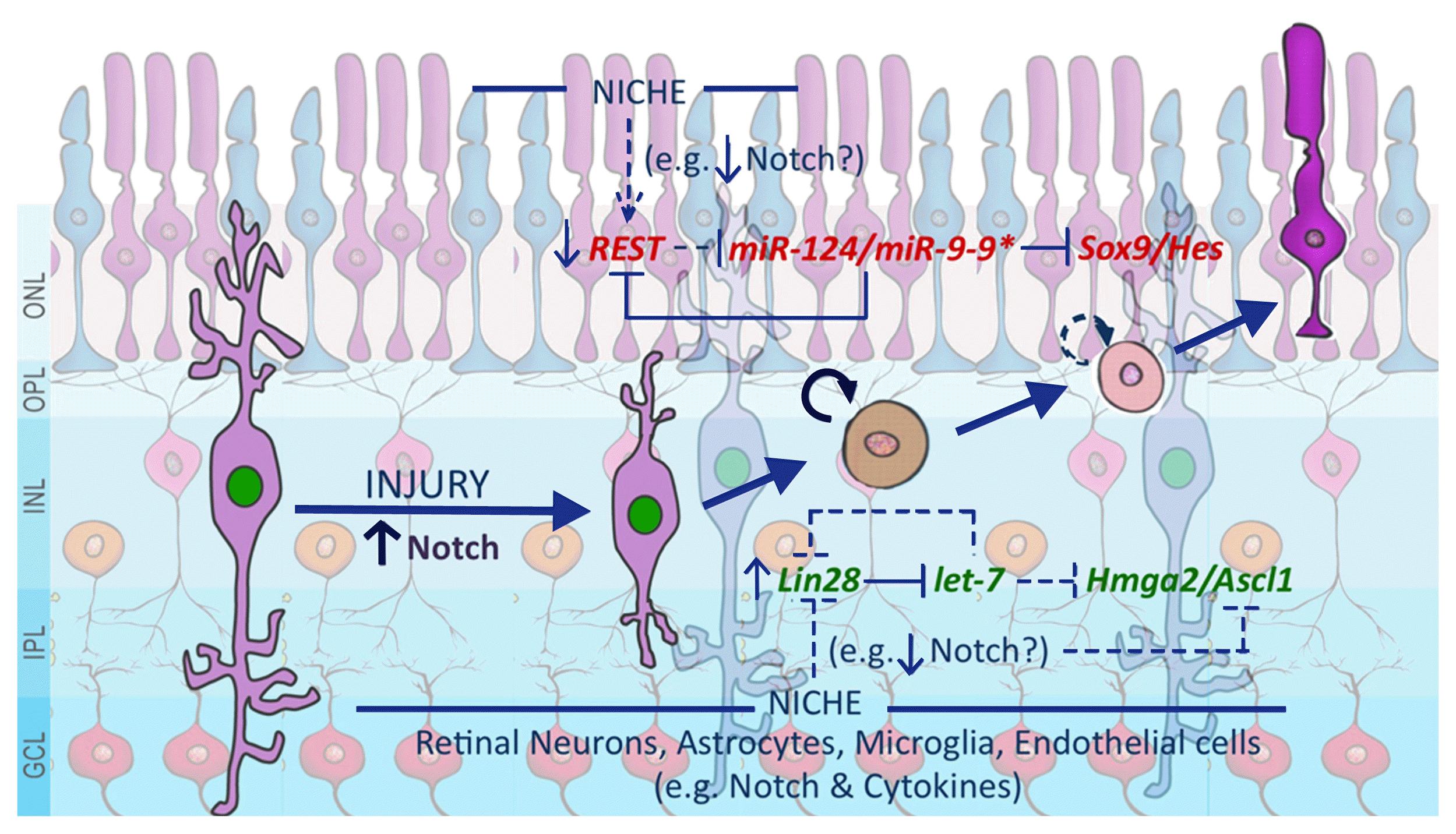 | Fig. 1A schematic representation of mechanisms underlying the neurogenic potential of MG. A subset of MG, most likely those with dormant stem cell properties, respond to injury by proliferating presumably under Notch signaling influence. As the activated MG migrate out of the INL, the changing niche, presumably reflected in altered Notch/cytokine signaling, influence them to engage both the excitatory (Green) and inhibitory (Red) axes regulating the neurogenic potential. It is possible that the imbalance between the two axes and their inadequate niche-based recruitment prevents mammalian MG from regenerating retinal neurons. The niche could be composed of retinal neurons, immigrant astrocytes, microglia, and endothelial cells. The niche-based communication for regeneration may involve diverse signaling pathways, exemplified by Notch and cytokine signaling, acting in concert. Notch signaling besides influencing these two molecular axes, may directly influence the expression of proneural genes. ONL: outer nuclear layer, OPL: outer plexiform layer, INL: inner nuclear layer, IPL: inner plexiform layer, GCL: ganglion cell layer.
|
Mechanisms underlying the neuroglial switch in the mammalian retina
The recent success of reprogramming to induce pluripotency in somatic cells and lineage-specific differentiation of pluripotent cells, whether through directed differentiation (SVZ/SGZ model) or trans-differentiation (Extra SVZ/SGZ model), is owed directly to the knowledge of developmental mechanisms (12). There is a significant knowledge gap in our understanding of how neurogenesis shifts to the generation of MG in the mammalian retina, information essential to navigate the cellular or molecular roadblocks to neuronal differentiation. For example, although we know that Notch signaling regulates the generation of MG, how it is incorporated into the gliogenic program and to what extent it is involved in suppressing neuronal differentiation remains rather unknown (4). Given the contextual role of Notch signaling (4), its complexity due to the oscillatory expression of its effector Hes1 (13), and its gliogenic interactions with the eye field gene Lhx2 (14), this information will be essential to formulate niche-based approaches for facilitating neuronal differentiation of MG. Along this line, it will also be advantageous to know whether genes involved in neurogliogenesis elsewhere in the CNS, such as Lin28, a heterochronic gene regulating the developmental timing (15) and Ezh2, an epigenetic regulator of gene expression (16) participate in MG differentiation and coordinate the influence of the niche. Recent observations that the decision of retinal progenitors during late histogenesis to differentiate along glial or neuronal lineage is influenced by Lin28 (17) and that Ezh2 plays a role in constraining the generation of MG (18) posit these genes as potential targets for pivoting MG torwards neuronal differentiation.
Molecular axes involved in MG-mediated regeneration
Regardless of whether or not MG possesses dormant stem cell properties according to the aforementioned models they must initiate and complete an intertwined program of activation and neuronal conversion for successful regeneration (4). Given that injuries in general lead to proliferation of MG across species, but not to their neuronal differentiation in higher vertebrates, it is likely that the cross-talk between transcriptional networks sub-serving the activation and neuronal differentiation are either not connected to the process, or the network components are in place but are not epigenetically primed for optimal expression in the mammalian MG. These probabilities could be examined by a genome-wide screening of prospectively enriched MG in select animal models in quiescent and activated states (facilitated by lineage reporters and other enrichment protocols such as the side population (SP) cell profiling by microarrays, RNA seq, and ChIP seq analyses. A similar approach to characterizing MG in different states of activation and differentiation in controlled conditions in vitro will yield corroborative evidence and facilitate the means to test putative mechanism(s) by manipulating gene expression. Together, these approaches may provide insight into molecular axes that can be tested against the background of those operational in zebrafish (7, 8, 19). For example, the molecular axis defined by Lin28 and Ascl1, which facilitates MG-mediated regeneration in zebrafish, is a valid target for study in mammals. A variety of approaches in higher vertebrates have demonstrated important regulatory roles for Lin28 in the maintenance of neural progenitors and neuroglial decision (20). It is likely that Lin28 influences different developmental function by contextual recruitment of Hmga2, a gene encoding a DNA architecture protein (21, 22) and Ascl1 (23) by directly regulating the heterochronic miRNA let-7 (17, 21, 23). let-7 targets Hmga2 (17, 21) and Ascl1 (23); therefore, the Lin28-let-7-Hmga2/Ascl1 axis has emerged as an evolutionary conserved axis that could be a candidate for unlocking neurogenic potential of MG. Early evidence supports this premise. For example, overexpression of Lin28a (24) in the enriched MG (Fig. 2A) prompted these cells to acquire neuronal morphology and to express immunoreactivities and transcripts corresponding to neuronal genes (Fig. 2B). More importantly, neuronal differentiation was accompanied by an increase in levels of miR-124, a proneural miRNA (25), Hmga2, and Ascl1, and a decrease in those of REST, a global inhibitor of neuronal differentiation (26), thus demonstrating the capacity of Lin28a in activating the neurogenic program in MG. However, targeting the Lin28-let-7-Hmga2/Ascl1 axis alone may not be sufficient for the functional reprogramming of MG along neuronal lineage, given that its influence may not be able to completely counter the inhibitory resistance of the REST axis. REST, by suppressing the expression of proneural miRNAs, miR-124 and miR-9-9*, whose targets are proglial genes encoding SOX9 and HES family of regulators, may keep MG non-neuronal and therefore non-regenerative (25). The existence of the REST-miR-124-9-9*-Sox9/Hes1 axis in MG suggested that either inhibiting REST or ectopically expressing miR-124/miR-9-9* or both may direct MG along the neuronal lineage. It is likely that the ectopic expression of miR-124/miR-9-9* in itself might be effective, as they directly or indirectly influence the expression and function of REST (25). This notion was supported by the following observations. For example, over expression of miR-124-9-9* (27) in MG (Fig. 2A) facilitated neuronal morphology with accompanied expression and suppression of neuronal- and glial-specific genes, respectively (Fig. 2C). Expression of both Ascl1 and Lin28a was up regulated and that of REST was down regulated in the perturbed groups, compared to controls. However, no significant difference in Hmga2 transcript levels was observed, suggesting a threshold requirement of Lin28a levels for influencing Hmga2 expression. That miR-124 and miR-9-9* could facilitate proneural function of Ascl1 was demonstrated by the improvement of Ascl1-mediated reprogramming of MG by ectopically expressed miRNAs (28). Together, these observations posit these molecular axes as valid intrinsic targets for studying and unlocking the dormant regenerative potential of MG.
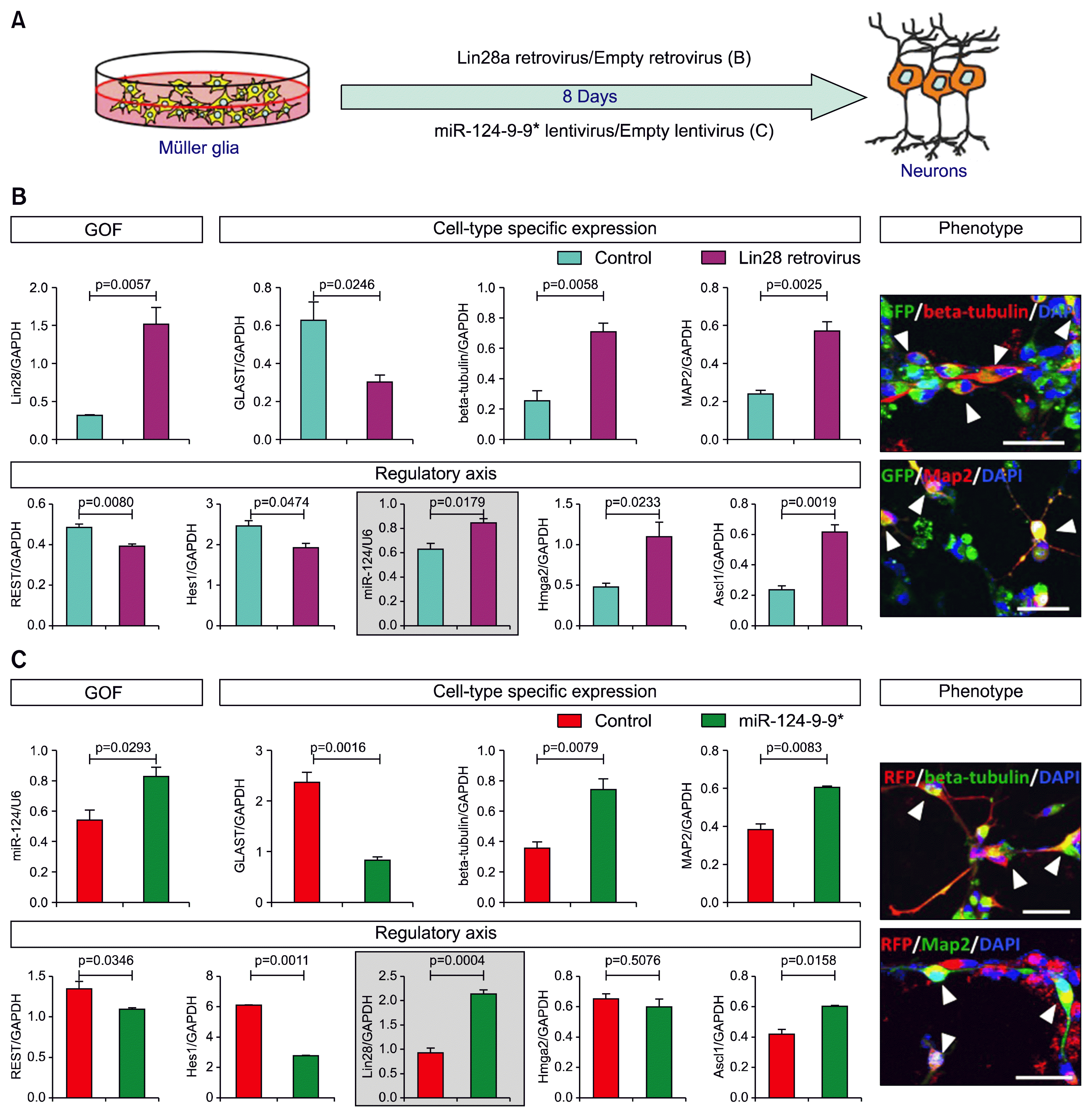 | Fig. 2The intrinsic molecular axes and their involvement in the neurogenic potential of MG. (A) A schematic diagram shows the virus-mediated ectopic expression in enriched MG to perturb Lin28a (Lin28a-let7-Hmga2/Ascl1) and miR-124 and miR-9/9* (Rest-miR-124/miR-9-9*-Sox9/Hes). (B) Lin28a overexpression caused a significant decrease and increase in transcripts characterizing MG (Glast) and neurons (beta-tubulin and Map2), respectively. Neuronal differentiation of transduced MG could also be ascertained by their neuronal morphology and expression of immunoreactivities corresponding to beta-tubulin and Map2. Neuronal differentiation of MG was accompanied by a significant increase and decrease in the expression of positive (miR-124, Hmga2, and Ascl1) and negative (REST and Hes1) regulators of neurogenesis. (C) Similarly, miR-124-9-9* overexpression led to a decrease in levels of glia-specific (Glast) and an increase in neuron-specific (beta-tubulin and Map2) transcripts. As observed in Lin28a overexpression, transduced MG displayed neuronal morphology and expressed beta-tubulin and Map2 immunoreactivities. A significant increase and decrease in the expression of positive (Lin28a and Ascl1) and negative (REST and Hes1) regulators of neurogenesis was accompanied by miR-124-9-9* mediated neuronal differentiation of MG. Levels of Hmga2 transcripts remained unchanged. That the two axes might intersect and influence each other was demonstrated by the increase of miR-124 transcript levels in Lin28 overexpression experiment and vice versa (enclosed graph, B, C). MG were enriched as described in Das et al., (6). They were transduced with Lin28a retrovirus (24) or miR124-9-9* lentivirus (27) and the effects of perturbations on gene expression and phenotype were examined by Q-PCR and immunocytochemical analyses, respectively, as described in Parameswaran et al., (22) and Xia et al., (17). Controls included MG transduced with empty retrovirus/lentivirus. Levels of transcripts and miRNA are presented after normalization with that of GAPDH and U6, respectively. Arrowheads indicate transduced MG (GFP+/RFP+) expressing neuronal markers, beta-tubulin or Map2. Scale bar, 50 μm. Data are mean±s.e.m.
|
Influences of the niche and how they are mediated
Regeneration takes place when homeostasis is disturbed. MG, the guardian of homeostasis in the retina, respond to injuries by proliferating and migrating out of the inner nuclear layer (5). However, despite proliferation and migration as in zebrafish retina, as mentioned, mammalian MG do not initiate regeneration effectively. This raises the possibility that, in addition to internal constraints discussed above, the environment in the mammalian retina might not be conducive for neurogenic conversion of MG. A niche-based approach to unlock neurogenic potential of MG will involve examining the relationship of MG with neighboring retinal cells, microglia, immigrant astrocytes, and endothelial cells in the context of signaling pathways and their capacity to engage the molecular axes involved in reprograming along neuronal lineage. For example, the understanding of the role of microglia, one of the first responders to injury, and which may mediate regeneration through inflammatory signals, is still evolving in the retina (29). Studies have demonstrated that Notch (8, 30), Wnt (5, 6), FGF (6, 8), insulin and insulin-like growth factor-1 (8), Shh (31), and cytokines such as TNFα (8, 19), leptin, and IL-11 (32) may play important roles in mediating the influence of the niche in reprogramming MG. However, the identity of cells delivering the signals and whether or not these signals and their associated pathways act in concert, which would determine niche-based strategies to promote regeneration, remain largely unknown. However, studies in zebrafish have begun to shed light on these issues. For example, it has been observed that TNFα released by dying photoreceptors in mechanically damaged zebrafish retina may constitute one of the early signals for activating MG for regeneration (19). Also, it was reported that zebrafish MG respond to retinal injury by secreting leptin and IL-11, which help reprogram cells in an autocrine fashion (32). In both cases, these cytokines facilitated injury-dependent induction of Ascl1, a key step in reprogramming of MG (8). Whether or not signaling-mediated by these factors are similarly involved in the mammalian retina remains to be demonstrated. Although the identity of cells delivering Notch signaling in MG remains speculative, emerging evidence from zebrafish and higher vertebrates posit it as an important niche-based target for MG-dependent regeneration. It remains active long after facilitating the differentiation of MG, enabling them to launch the proliferative response when injury takes place (4, 5). However, persistence of Notch signaling in activated MG, which are poised for neuronal conversion, might be deleterious for regenerative process, given the inhibitory influence of Notch signaling on the activation of proneural genes (33). The inability to confine the activation of Notch signaling in MG to a narrow temporal window following injury may be one of the reasons for their anemic neuronal conversion in mammals, compared to that in zebrafish (30). Emerging evidence, once again from the zebrafish model, suggests that these pathways, including Notch signaling, may act in concert and MG integrate the diverse niche-based influence of a variety of signaling molecules for a calibrated regenerative response to injury (8). Therefore, niche-related information is crucial for understanding the temporal and spatial modulation of signaling required for activating MG and shifting them from the state of activation to neuronal differentiation.
Markers of activated MG
This is fundamental information missing from studies of MG-dependent regeneration, without which the underlying mechanisms remain difficult to grasp and the therapeutic target rather illusory. Currently, the identity of the activated MG represents cells in the inner nuclear layer of the retina, which have incorporated BrdU in response to injury and express MG-specific markers. Given the transition of MG from a quiescent to a proliferative state and the generation of their progenies in different stages of development, the identity based on BrdU incorporation, though important, is of limited value. Attempts have been made to prospectively enrich activated MG by Hoechst dye efflux assays and to characterize them as SP cells, a phenotype shared by the majority of stem cells (4, 6). However, since SP cells are also heterogeneous, this functional approach has limitations in the absence of specific cellular markers. Although several signaling pathways (e.g. Notch signaling) and intrinsic factors (e.g., Ascl1) have been observed to regulate MG activation in fish, birds, and rodents (8), their status as markers of activated MG in different stages of regeneration remain unspecified. Progress in understanding the regenerative mechanisms in tissues like blood (34), intestine (35), and skin (36) has been facilitated by the identification and characterization of markers of the resident stem cells and their progenies. Within the CNS, insight into regeneration in the SVZ has come from the characterization of B1 quiescent and active progenitors and resulting neuroblasts (37). Therefore, to follow MG-mediated regeneration, spatially and temporally, and to identify the stage-specific intrinsic players, a concerted effort is needed to search for reliable and reproducible markers.
Animal models for MG-mediated therapeutic regeneration
Currently, there is no consensus on reliable and reproducible mammalian models to study the activation and neurogenic potential of MG. Rodent models representing retinal injuries, ranging from those caused by the exposure to light, neurotoxins, and genetic mutations have been used. Given the observations that glia respond differently to different types and durations of injuries (2), that their levels of activation differ from species to species (Ahmad et al., unpublished observations), and that within the same species there are strain differences in responses (38), a case may be made for developing injury- and species/strain-specific models for reproducible and unambiguous examination of MG neurogenic potentials. These models will be valuable in incorporating transgenic technology for reliable lineage tracing, and more importantly for molecular characterization of activated MG. Characterization of these cells for gene regulatory network and epigenetic signature is essential to shed light on the status of their neurogenic potential and approaches to unlock it. Furthermore, cell type specific injury models would test the ability of MG to replace specific neuronal types, which will be helpful in designing approaches for MG-dependent therapeutic regeneration. For example, the loss of vision in two of the intractable blinding diseases, age-related macular degeneration (AMD) and glaucoma, is due to selective loss of photoreceptors and retinal ganglion cells (RGCs), respectively. The neurotoxin injury models where exposure of the retina to N-Methyl-N-nitrosourea (MNU) ablates photoreceptors (39) and NMDA causes the degeneration of RGCs (40) would reveal whether or not MG could differentiate along specific neuronal types. Also, it may shed light on if MG-dependent therapeutic regeneration would be a practical approach to address degenerative changes in AMD and/or glaucoma.
Go to :
Conclusions
The discipline of MG-based regeneration is a recent one, particularly in mammals. With information on the nature of the activated MG and the molecular axes mediating their response to the niche, aided by reproducible animal models and lineage reporters, a pharmacological recruitment of these endogenous progenitors for therapeutic regeneration is a near possibility. In addition, information emerging from these studies will help us understand how the neurogenic potential in the adult mammalian brain is constrained relative to that of lower vertebrates, currently a significant knowledge gap.
Go to :
 XML Download
XML Download