

 PDF
PDF Citation
Citation Print
Print


Introduction
Neural lineage differentiation in vitro using pluripotent stem cells (PSCs) has now progressed beyond the differentiation into simple neurons and glial cells, to the creation of organ-like tissues, or organoids (1-4). Human brain organoids contain various cell types such as neurons, glia, and microglia that constitute brain tissue, and their structure even resembles some regions of the actual human brain (5, 6). Therefore, brain organoids are the best tool to investigate the interaction between the neurons and supporting cells in the brain. The traditional two-dimensional (2D) cultures have limitations in mimicking the complexities of cell-cell interactions, 3D cell patterning, and cellular properties of the human brain (7-9). During the differentiation of PSCs, including human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), the differentiating cells are spatiotemporally self-assembled and spontaneously form natural tissue-like structures (7, 10).
Neural lineage differentiation from human PSCs, starting with neural rosette formation using embryoid bodies, was reported in 2001; the development of cerebral brain organoids from human PSCs was first reported in 2013 (5, 11). Lancaster et al. embedded the embryoid bodies committed to a neuroectodermal lineage into Matrigel droplets to induce 3D tissue growth and used a spinning bioreactor to evenly provide nutrients and gases during long-term culture (5, 12). Furthermore, Kadoshima et al. found that human ESC aggregates spontaneously formed dorsocaudal-ventrorostral axis with cortical tissues (13). In the present study, we developed an optimized methodology for the generation of cerebral organoids using hESCs and hiPSCs, based on Lancaster’s protocol (12).
Go to : 
Materials and Methods
Cell cultures
For feeder-free cultures, hESCs (National Stem Cell Bank, Republic of Korea) and hiPSCs were cultured on a vitronectin-coated tissue culture dish in TeSR-E8 medium (Stem cell technologies, VBC, Canada). The PBS-diluted vitronectin (Thermo Fisher, MA, USA, 5 µg/ml) was added to the tissue culture dish and incubated at 20∼25℃ for 1 h. Cells were sub-cultured every 4∼5 days in a 0.5 mM EDTA solution (Enzynomics, Daejeon, Republic of Korea), and 10 μM Y-27632 (Peprotech, Rocky Hill, USA) was added on the day of subculture.
Cerebral organoid generation from feeder-free cultured hPSCs
The cerebral organoid generation protocol was slightly modified from that reported previously (12), particularly in the early stages. The hESCs and hiPSCs were dissociated into small clumps with a 0.5mM EDTA solution and plated on suspension dishes containing the Aggrewell EB formation medium (Stem Cell Technologies, VBC, Canada) and 10 μM Y-27632 for the first 2 days. The aggregated EBs were then washed every other day with TeSR-E8 medium supplemented with penicillin/strepto-mycin (Gibco, TX, USA). After 4 days, they were transferred to neural induction medium (DMEM/F12 [Gibco] supplemented with 1:100 N2 supplement [Gibco], 1% nonessential amino acids [Gibco], 1× penicillin/strepto-mycin/glutamine [Gibco], and heparin 1 μg/ml). The EBs were then washed every other day with fresh neural induction medium until a transparent border was observed. The resulting EBs were then embedded into a Matrigel (BD biosciences) droplet on a parafilm sheet with dimples and incubated at 37℃ for 40∼60 min to solidify the Matrigel. The parafilm sheets were removed, and the embedded EBs were cultured for 6 days in cerebral organoid differentiation medium (a 1:1 mixture of DMEM/F12 [Gibco] and Neurobasal [Gibco] medium supplemented with 1:200 N2 supplement [Gibco], 1:100 B27 without vitamin A [Gibco] 1% nonessential amino acids [Gibco], 1× penicillin/streptomycin/glutamine [Gibco], 2-mercaptoethanol 0.5 μM [Gibco], and insulin 2.5 μg/ml). The EBs were then cultured in cerebral organoid maturation medium (a 1:1 mixture of DMEM/F12 [Gibco] and Neurobasal [Gibco] medium supplemented with 1:100 N2 supplement [Gibco], 1:50 B27 [Gibco] 1% nonessential amino acids [Gibco], 1× penicillin/streptomy-cin/glutamine [Gibco], 2-mercaptoethanol 0.5 μM [Gibco], and insulin 2.5 μg/ml). The detailed sequence of hESC differentiation and the culture of cerebral organoids is described in the Results section.
Microsection and immunocytochemistry
The cerebral organoids tissues were fixed with 4% paraformaldehyde (Sigma, St. Louis, MO, USA) for 2 h at 4℃. For cryosectioning, the organoids were incubated in a 30% sucrose solution in PBS for 2 days at 4℃ until they sink. Tissues were then embedded in OCT compound (Tissue-Tek) and frozen at −80℃. A Cryotome (Leica, CM-1850) was used for preparing sections, and tissues were sectioned at −25℃ with 20 μm thickness. After sectioning, the tissues were washed with PBS (Gibco) for 30 min to eliminate the residual OCT compound. For vibratome sectioning, the fixed organoids were embedded in 3% agarose gel. Slicing was performed using a 7000 SMZ vibratome (Campden) in PBS at 0.3 mm/s speed, 1.00 mm amplitude, and 50HZ. Then, 50∼100 μm cerebral organoid slices were peeled off at a time from the sample and gathered using a small art brush. The sectioned tissues were treated with PBS, containing 3% bovine serum albumin and 0.03% Triton X-100 (Sigma), for 45 min at 20∼25℃.
The adherent cells were fixed with 4% paraformaldehyde (Sigma) for 20 min at 4℃. After washing with PBS (Gibco), the cells were treated with PBS, containing 3% bovine serum albumin and 0.03% Triton X-100 (Sigma), for 45 min at 20∼25℃.
For 3D immunocytochemistry, cerebral organoids were fixed with 4% paraformaldehyde (Sigma) for 1 day at 4℃. After washing with PBS (Gibco), the tissues were treated with PBS, containing 6% bovine serum albumin and 0.03% Triton X-100 (Sigma), for 1 day at 20∼25℃. Primary antibodies and fluorescence-labeled secondary antibodies were used according to the manufacturer’s speci-fications. The primary antibodies used were anti-OCT4 (OCT4; polyclonal, 1:500, Santacruz), anti-TRA-1-81 (TRA-1-81; monoclonal, 1:500, Santacruz), anti-SOX2 (SOX2; polyclonal, 1:500, Millipore), and anti-TUJ-1 (TUJ-1; polyclonal, 1:300, BioTechne). For detecting the primary antibodies, fluorescence-labeled (Alexa Fluor 488, 568, or 647; Molecular Probes, Eugene, OR, USA) secondary antibodies were used according to the manufacturer’s specifications. For organoid clearing, cerebral organoids were immersed in the clearing solution (25% urea and 65% sucrose in H2O), and the cerebral organoids were observed to become transparent for up to 20 mins.
Go to :
Results
Selection and culture of human pluripotent stem cells
Two types of human PSCs (hPSCs), hESCs and hiPSCs, were generally used for generating cerebral organoids. For cerebral organoid formation, hESCs and hiPSCs were routinely maintained on a vitronectin-coated culture dish with animal component-free TeSR-E8 medium. Every 4∼5 days, cells were passaged as small clumps using 0.5 mM EDTA, and the medium was replaced with fresh medium every day. Choosing appropriate hPSCs is the first step for generating cerebral organoids, as every cell has a different differentiation propensity. We checked whether the hESCs and hiPSCs had a round and flat morphology with a normal growth rate (Fig. 1A and 1C), and whether the feeder-free cultured hESCs and hiPSCs expressed the pluripotency marker OCT4, TRA-1-81, and SOX2 (Fig. 1B).
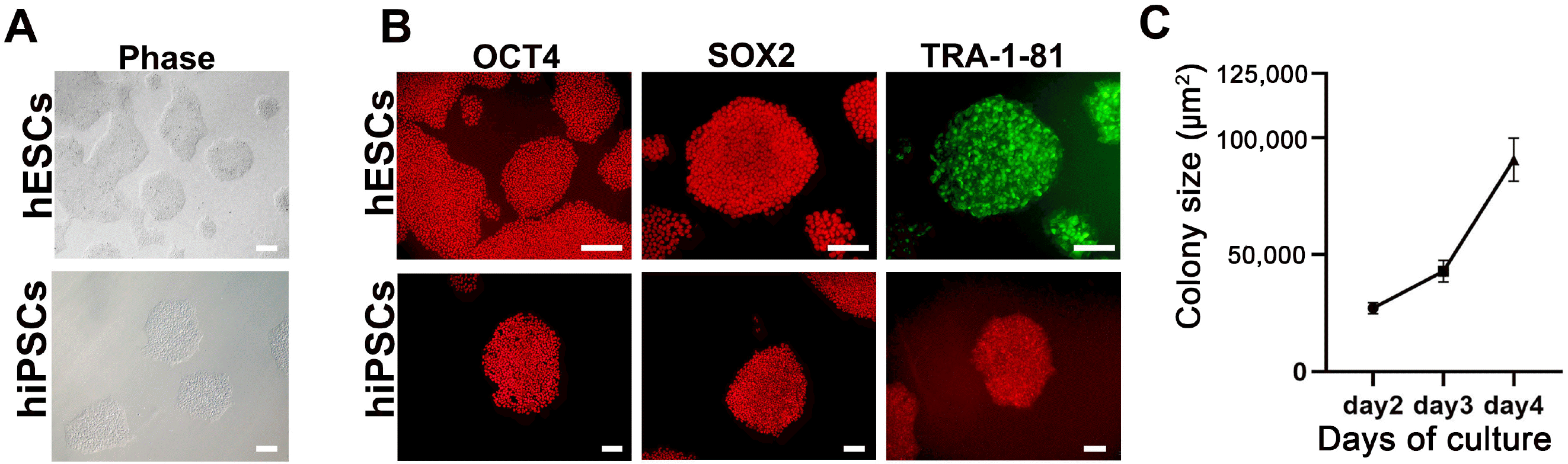 | Fig. 1Characterization of human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs). (A) Phase contrast images of hESCs and hiPSCs in feeder-free conditions. Scale bar=200 μm. (B) Immunocytochemical staining of the pluripotency markers OCT4, SOX2, and TRA-1-81 in hESCs (upper) and hiPSCs (lower). Scale bar=200 μm in the upper panels and 100 μm in the lower panels. (C) Growth rate of hESCs from 2 to 4 days after seeding. Growth rate was measured as an increase in colony size (μm2). Error bars represent standard error of the mean.
|
Neural lineage induction via embryoid body (EB) formation from hPSCs
The schematic protocol for cerebral organoid generation is summarized in Fig. 2A. For the embryoid body (EB) formation, small dissociated clumps of hESCs and hiPSCs were seeded with AggreWell EB formation medium on the Petri dish, and 10 μM of Y-27632 (ROCK inhibitor) was added to increase the cell survival rate. Although 20% knockout SR-containing medium was sufficient to form EBs from hPSCs, the AggreWell EB formation medium could support EB formation from cell lines even with poor EB formation ability. The AggreWell EB formation medium could produce more homogenous and rapidly proliferating EBs compared to knockout SR-containing medium (Fig. 2C). On day 2 of differentiation, the aggregates were washed and transferred to new Petri dishes with TeSR-E8 medium for EB growth. EBs were then washed with the TeSR-E8 medium every 2 days until they reached a size of 300∼500 μm. Through this process, EBs underwent early neural specification at about day 6∼8 of aggregate culture (Fig. 2B). The neural lineage-committed EBs were then washed and transferred into new Petri dishes with neural induction medium (NIM) containing N2 supplement. The EBs were washed every other day with fresh NIM. As the neural specification progressed, outer surface of the EBs formed a distinct transparent layer with a neuroepithelial-like structure. Under a light microscope, we could observe bright and transparent monolayers, which resembled the neuroepithelium of the cerebral cortex (Fig. 2B). In general, EBs differentiated into neuroepithelium-containing structures by day 4∼6 of neural induction, when they exhibited sufficient neural properties to proceed for further differentiation. If neuroepithelium layer formation is delayed during differentiation, EBs can be cultured in NIM for about 2 weeks. EBs start to have distinct layers with dark middle and bright edges that are transparent.
 | Fig. 2Brain organoid differentiation protocol using human pluripotent stem cells (hPSCs). (A) Schematic illustration of the generation of brain organoids. Representative images characterizing each stage are shown. Scale bar=200 μm. (B) Morphology of embryoid bodies (EBs) in EB formation medium, based on TeSR medium (upper left), and their enlarged images (upper right). EBs committed to the neural lineage showed a transparent neuroepithelium-like region at the periphery of the structure (lower left), which is depicted with a white dotted circle (lower right). Scale bar=500 μm (lower left), 200 μm (lower right). (C) Comparison of koSR-containing medium (hESC medium) and Aggrewell EB medium for the size of EBs differentiated from hESCs. Scale bars=200 μm.
|
Embedding of EBs into Matrigel and subsequent culture for cerebral organoid formation
To use Matrigel, it must be liquefied at 4℃ by refrigerating one day before use. After washing with fresh medium, EBs with a transparent border were selected and embedded in Matrigel droplets on a dimpled parafilm sheet (Fig. 3A) (12). Matrigel containing EBs was then solidified at 37℃ for about 40∼60 min in an incubator. After solidification, the EB-containing Matrigel was transferred into a Petri dish and cerebral organoid differentiation medium was added. If a lot more organoid samples were required, embedding was performed in a different manner (Fig. 3A). First, 150 μl of liquefied Matrigel was spread in a 4-well dish and allowed to solidify for about 30 min. Then, 10∼15 EBs were selected and evenly distributed on the solidified Matrigel, followed by covering with 150 μl of Matrigel; the EBs were thus sandwiched between two Matrigel sheets. After solidification, 1 ml of cerebral organoid differentiation medium was added. The medium was changed every other day, taking care to avoid the breakage of Matrigel. On day 6 after Matrigel embedding, the cerebral organoid differentiation medium was substituted with cerebral organoid maturation medium. At this stage, the neuroepithelium-like surface region of EBs was folded and neural tube- and neural rosette-like structures were formed in the inner part of the EBs. These structures were continuously generated anew during the culture (Fig. 3B). Even on day 4 of Matrigel embedding, high-contents screening (HCS) and 3D staining facilitated the visualization of neural tube structures in the inner part of the EBs (Fig. 4C). Finally, circular-shaped EBs were transformed into the early-stage cerebral organoids through a neural folding process.
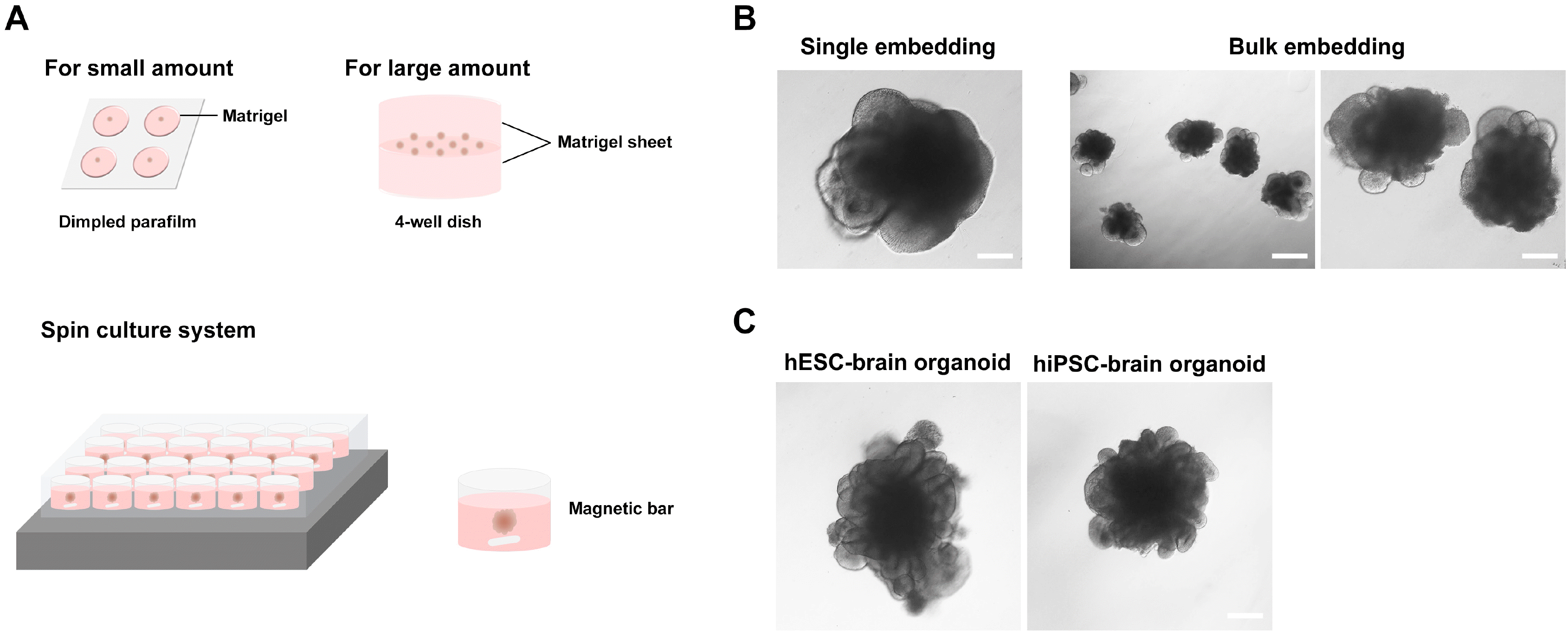 | Fig. 3Matrigel embedding culture and morphology of early-stage brain organoids. (A) Matrigel embedding process for small or large amount of embryoid bodies (EBs) and the spin culture system. (B) The neural folding stage of hESC-derived EBs at day 6 after Matrigel embedding with single (small amount) or bulk (large amount) EBs. Scale bar=200 μm (left and right) and 500 μm (middle). (C) Brain organoids with multiple neural rosette-like structures derived from human embryonic stem cells and (day 19) and human induced pluripotent stem cells (day 20). Scale bar=500 μm.
|
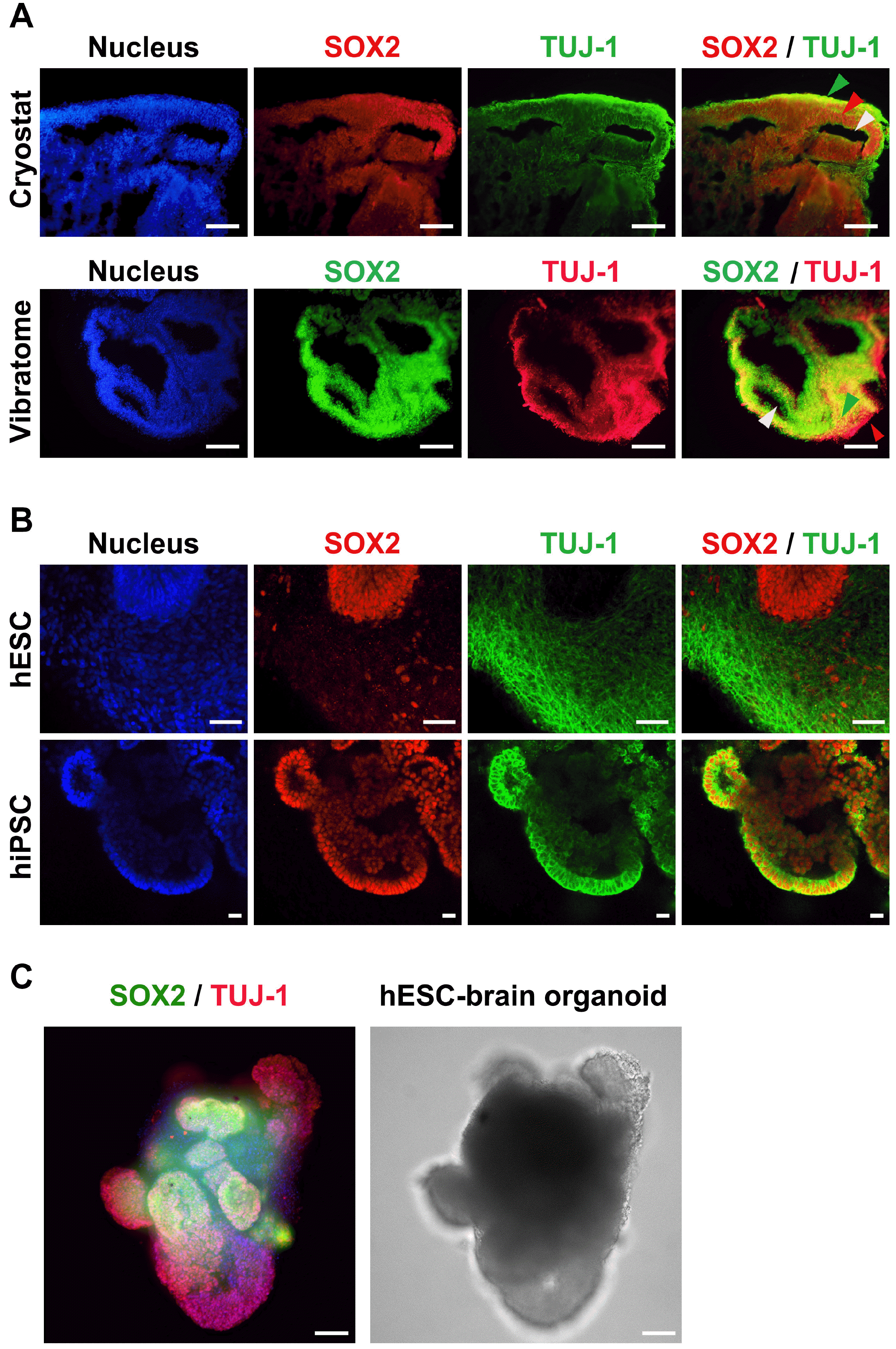 | Fig. 4Characterization of human embryonic stem cell (hESC)- and human induced pluripotent stem cell (hiPSC)-derived brain organoids. (A) 2D immunocytochemistry staining of the early neuron marker TUJ-1 and the neural progenitor marker SOX2 in hPSC-derived organoids. A Cryostat (upper) and Vibratome (lower) were used for tissue sectioning. Scale bar=100 μm (upper panels) and 200 μm (lower panels). Green arrow indicates cortical plate, red arrow indicates ventricular zone, and white arrow indicates ventricle (Cryostat sample). Green arrow indicates ventricular zone, red arrow indicates cortical plate, and white arrow indicates ventricle (Vibratome sample). (B) Confocal images of 3D immunocytochemistry for the early neuron marker TUJ-1 and the neural progenitor marker SOX2 in hESC- and hiPSC-derived organoids. Scale bar=50 μm (upper panels) and 20 μm (lower panels). (C) Representative 3D image of whole brain organoids, which were stained with TUJ-1 and SOX2 (at day 12 hESC-derived). The image was captured using high contents screening (HCS). Scale bars=200 μm.
|
The major limitation of the current brain organoid culture methods is the apoptosis or necrotic body formation in the internal parts as organoids grow in size during long-term culture. In the living body, the vascular system is spread throughout the organs to provide a continuous supply of oxygen and nutrients to the deeper parts of the body. However, in vitro organoid culture systems cannot supply oxygen and nutrients to the internal parts of organoids due to a lack of vascular formation. To address this problem, many laboratories use an agitation culture system, or attempt to culture organoids as small pieces (5, 14-16). To develop a cost-effective method for organoid culture, we used a magnetic spin culture system (Fig. 3A). On day 6 after embedding in Matrigel, cerebral organoids were transferred into a 24-well dish containing 2 ml cerebral organoid maturation medium, with a mini magnetic bar at the bottom of each well. Each mini magnetic bar can be controlled independently by using 2mag Magnetic stirrer (Germany). Using magnetic spin culture system, 24 organoids can be individually cultured in each well of a 24-well dish. Although the Matrigel surrounding the organoid is slightly damaged by the rotating magnetic spin bar, this system is sufficient for culturing the organoids up to 2∼3 mm in diameter. For up to 40 days, cerebral organoids can be cultured in a non-rotating system. However, spin culture system could be effective for long-term culture, as internal necrosis usually begins to occur at approximately 30∼40 days of organoid culture (from EB formation).
Characterization of cerebral organoids
Approximately 30 days after embedding in Matrigel, the cerebral organoids showed typical characteristics, such as a folded neuroepithelium like-surface, a large number of rosette structures, and formation of ventricular zones. Thus, the normal formation of early cerebral organoids could be confirmed via light microscopy, through which the following features were checked: 1) a folded structure on the outer surface, 2) vertically arranged cells in neuroepithelium-like region, and 3) the existence of rosettes and ventricular structures in the interior (Fig. 3C). To confirm the normal formation of cerebral organoids, further analyses are required. We determined whether the organoids were composed of neural lineage cells, and which part of the brain tissue they were close to. Transcriptome analysis only shows whether cerebral organoids contain neural lineage cells. Therefore, to determine how closely organoids mimic the brain structure, immunocytochemistry analysis is an essential approach for organoid characterization. The detailed structure of cerebral organoids can be observed in 2D and 3D images using confocal microscopy. To obtain 2D images, sectioned tissues were immunostained with several neural markers, and images were then captured using a confocal microscope (Fig. 4A). To section organoid tissues, two methods were used: cryosectioning and vibratome sectioning. Both methods have their pros and cons; cryosectioning using a cryostat can cut tissues to a thickness of 20 μm whereas the lowest section thickness that a vibratome can reliably achieve is approximately 50 μm. However, cryostat-cut sections often show tissue tearing, whereas vibratome sections are relatively defect-free. When images are acquired using a confocal microscope, this slight difference in thickness does not affect the image. Thus, vibratome sectioning is recommend because this method does not require dehydration and freezing and causes less tissue damage. To obtain 3D images, CLARITY is required for tissue clarification to make the tissue transparent before immunostaining the cerebral organoids using the clearing solution (17, 18). The 3D images can reveal the overall shape and structure of a whole organoid, whereas 2D images can provide the precise structure of the internal regions of organoids. Therefore, we recommend performing both 2D and 3D image analyses to accurately identify the characteristics of the organoids. As the hierarchical arrangement of different cell types in cerebral organoids is the most prominent feature of the cerebral cortex, we stained both a progenitor marker (SOX2) and neuronal markers (TUJ1) in the same sample. We could observe an ventricular zone (VZ), and a cortical plate (CP) with developing neurons (Fig. 4A and 4B) in the hESC-derived cerebral organoid. The hiPSC-derived cerebral organoid showed a relatively indistinct layer distribution; however, vertically arranged oval-shaped cells co-expressing SOX2 and TUJ-1 were present in the neuroepithelium-like region, which may represent the neural progenitors differentiating into neurons. The cortical regions of hiPSC-derived cerebral organoids appear to be relatively immature and may require further cultivation for neuronal differentiation (Fig. 4B). In general, hiPSCs are differentiated more slowly than hESCs. Further, the differentiation rate may vary slightly from cell line to cell line; thus, the duration of each organoid differentiation stage should be adjusted according to the cell lines and their status. Most cells constituting the 30-day-old brain organoids are neurons and neural stem cells, and there are almost no glial cells, such as astrocytes and oligodendrocytes. This is because glial specification in brain organoids usually occurs after culture for more than 100 days (19, 20).
Go to :
Discussion
Recently, with the success of clinical studies and cell therapy application cases using iPSCs (21), the clinical applicability of iPSC-derived brain organoid has increased. To take one step closer to clinical application or drug screening using hiPSC-derived organoids, an efficient protocol that can be applied to various types of patient-derived iPSCs is needed. To date, the routine formation of brain organoids using hPSCs with a single standardized protocol has not been reported. This is because each hPSC line displays different properties and, sometimes, even the same cell line shows a different cell state at the time of differentiation. Cells are sensitive to the environment depending on subculture density, experimental skills, and incubating condition. Therefore, the protocol presented in this study may need to be modified to suit each laboratory even if the same hPSC line is used. For cerebral organoid production, a thorough control of these variables is needed to maintain the optimal state of hPSCs. Furthermore, it is necessary to minimize heterogeneity during the subsequent differentiation process. Accordingly, we attempted to develop an optimal differentiation protocol for generating cerebral organoids with minimal protocol changes between using hESCs as well as hiPSCs. We shortened the EB formation time and enhanced EB formation efficiency using an aggregation-promoting medium. Cerebral organoid differentiation begins with cell clumps, as they are prone to death when treated as single cells. Thus, cell numbers need to be adjusted to form EBs with a uniform size as the different cell numbers can make large difference in the size of EBs, which cause heterogeneity in each cerebral organoid. When homogenous organoids are used for drug screening, it is possible to reduce the error in the effect of the treated drug. Another important issue to consider is the complete removal of undifferentiated cells and cells of other lineage. For this, dual SMAD inhibition, which blocks differentiation into the mesoderm and endoderm, may be applied at the beginning of hPSCs differentiation, or a method using human neural progenitor cells (hNPCs) other than hPSCs could also be considered (14, 22, 23). To eliminate undifferentiated cells from the differentiating/differentiated population, fluorescence activated cell sorting after PSC-specific surface maker staining is feasible. A recent study used quercetin to selectively remove undifferentiated cells with 99.99% efficiency; this method can also be applied clinically (24). Further, necrosis at the center of maturating brain organoids is a major challenge that limits long-term culture and further applications. A live section method could be one of the solutions to increase the survival rate and maturity of the organoids, enabling their long-term culture (16). By using vibrating microtomes, such as the Vibratome or Compress-tome, live organoids could be divided into small pieces, with the same thickness, that maintain their viability and maturity with minimum damage. These sectioned organoid fragments can be recultivated for the further maturation of organoids, and some of these fragments can be used for quality control and characterization.
One of the obstacles in brain organoid generation is the delayed glial specification during brain organoids formation, which may limit the value and clinical applicability of brain organoids, as it usually takes more than 100 days (19, 20). However, recent studies have suggested methods to promote glial and microglial differentiation within cerebral organoids. Shaker et al. developed a protocol to generate human brain organoids containing oligodendrocytes, within 42 days of differentiation. The cocktail of growth factors and small molecules, including thyroid hormone T3, neurotrophin NT3, hepatocyte growth factor, insulin growth factor and platelet-derived growth factor, could drive the differentiation of oligodendroglial lineage in brain organoids (25). Furthermore, Ormel et al. (26) found that microglia-like cells, which were derived from the mesoderm lineage, were present in cerebral organoids. The microglial marker IBA-1 was examined from as early as day 24 of differentiation and IBA-1+ cells were localized throughout the organoids at later stage.
Ultimately, the generated brain organoids should have uniform properties and functionality that meets the minimum standards; the general criteria for determining qualified brain organoids should also be established. As many researchers investigate this topic intensively, brain organoids could be a promising source for drug screening and tissue replacement therapy using the patient’s own 3D brain structure.
Go to :
 XML Download
XML Download