

 PDF
PDF Citation
Citation Print
Print


Introduction
The brain is undoubtedly the most sophisticated and vital organ of the human body. The development of human brain is complex and is dictated by large set of molecular events that are tightly regulated in a spatio-temporal manner. A slight alteration to this regulation in the fetal brain, results in various kinds of neural disorders, collectively referred to as neurodevelopmental disorders (NDD). Most of the NDDs result from the mutations/deletions in a gene or a collection of genes or a chromosomal location that are required for normal brain development. The other contributing factor to NDD is the environment, such as maternal use of alcohol, viral infection during pregnancy.
The prevalence of NDD in the United States has been increasing significantly since 2006 (1). There is no effective treatment for the reversal of any of the NDD. Current treatment modalities include administering drugs to delay disease progression and rehabilitation therapies. Due to the lack of knowledge of the complete set of molecular events that govern brain development, most of the NDDs are not completely understood.
Our understanding of vertebrate brain development and its malfunction have come from vertebrate animal models and the discarded human fetus. While animal models have been excellent tools to dissect out various genes responsible for various processes in brain development, generating and analyzing the animal models are time consuming and laborious. Importantly, they are not human and do not completely mimic the human brain development as the human brain has undergone remarkable changes, particularly cortical expansion, compared to other vertebrates and to even non-human primate species. With the advancement in stem cells technologies, especially the human embryonic stem cells (hESCs) (2) and human induced pluripotent stem cells (iPSCs) (3), human developmental biology and genetic disease modeling have taken a complete turn. Almost all types of neural cells, including but not limited to neural stem cells, neurons, retinal cells, astrocytes, oligodendrocytes and microglia, can be derived from pluripotent stem cells (PSCs) following developmental principles (4, 5). Disease specific iPSCs (6) have been used to obtain neurons, oligodendrocytes and microglia to model various neurological diseases (4).
The intrinsic ability of the PSCs to self-organize and form the polarized three dimensional (3D) cortical tissue under serum free conditions and extrinsic signals was first demonstrated by the Sasai group (Fig. 1) (7). Afterwards, multiple research groups have established different protocols by modifying the early developmental cues (majorly WNT, FGF, retinoic acid (RA) and TGFβ/BMP signaling) to form different types of human brain organoids. The brain organoid techniques that use the extrinsic signaling factors are broadly called “guided” methods. Lancaster et al. (8) further demonstrated that brain organoids, referred to as cerebral organoids, can be obtained without extrinsic signals. This methods is called, “unguided” approach (Fig. 1). Lancaster et al, focused on improving growth conditions by providing the environment necessary for intrinsic cues to influence development and avoided the use of patterning growth factors or other cell signaling agonists and antagonists. Briefly, they used Matrigel, an extracellular matrix (ECM) supplement, to provide structural support for the formation of organoids, and a shaker/agitator along with the growth medium to support the diffusion of oxygen and nutrition to the inner side of the organoids. This method resulted in cerebral organoids with various discrete, but interdependent brain regions of CNS – forebrain, midbrain and hindbrain (8), and exhibited gene expression patterns similar to the developing fetal cerebral cortex (9). It is important to note that the cerebral organoids obtained by the unguided method have a high degree of organoid to organoid variability in 1) cellular composition, 2) reproducibility and 3) differentiation efficiency (8-11). Furthermore, these organoids contain a diverse pool of neurons, glia, photosensitive cells and even non-ectodermal cells (11). The cellular diversity and variability in these organoids make them non-ideal for specific neuronal disease modeling and drug screening. Brain organoids for modeling various neurological diseases including NDD has been extensively reviewed earlier (12-14). Table 1 lists studies on NDD using brain organoids from either guided or unguided methods (15-37). In this review, we focus on the various NDDs modeled using the region-specific brain organoids from guided methods.
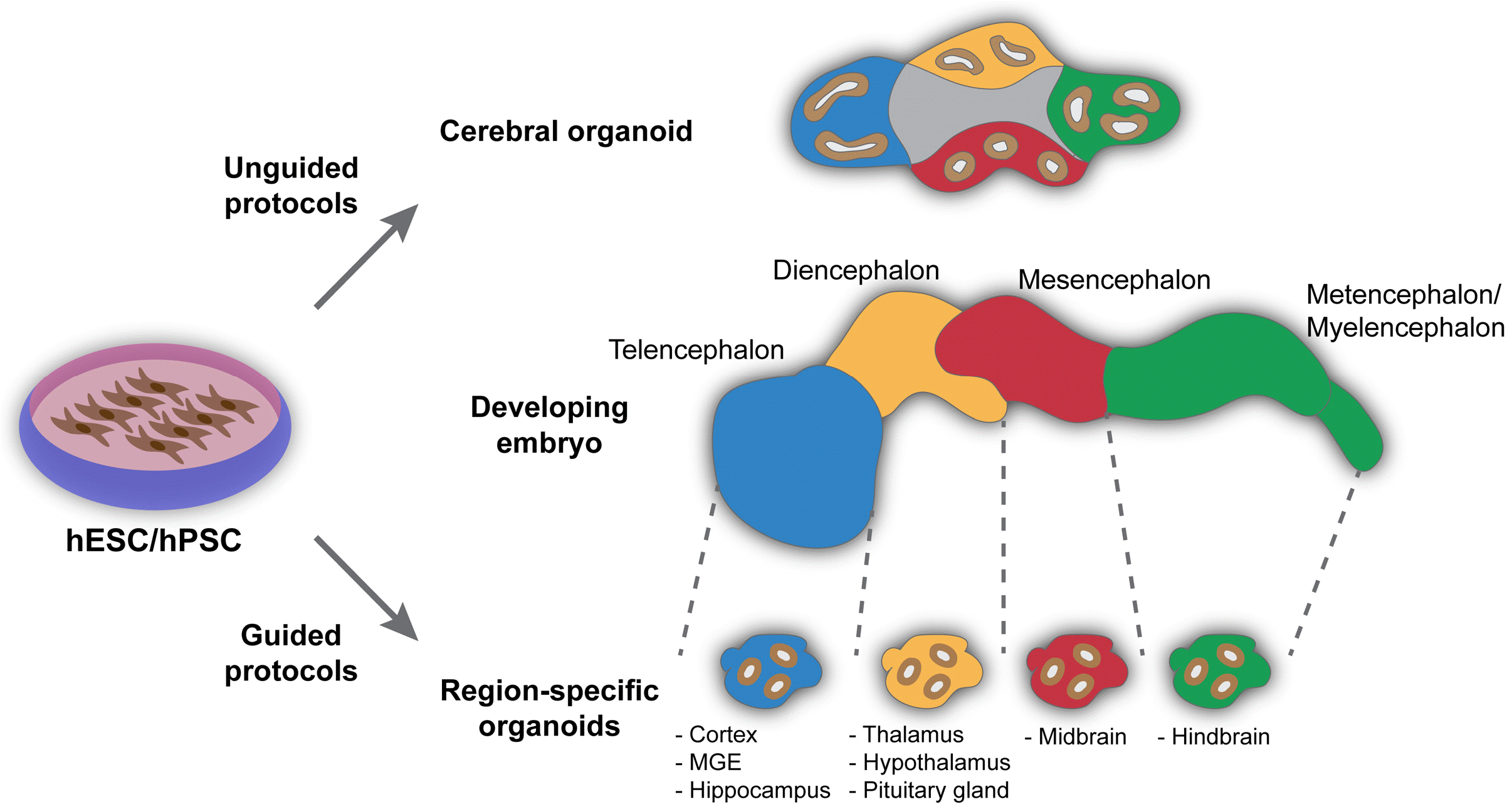 | Fig. 1Approaches for organoid ge-neration. Unguided approach: Orga-noids are developed based on self-organization principles of stem-cell aggregates resulting in cerebral orga-noids. Cerebral organoids contain heterogeneous population of tissues resembling various embryonic brain vesicles (middle panel). Guided approach: Stem-cell aggregates can be directed to distinct cell fates resulting in region-specific brain organoids in the presence of external patterning cues.
|
Table 1
NDDs and other neural disorders modeled so far using cerebral organoids
| Neurodevelopment disorder modeled | Modeled gene mutation/environmental condition |
Patient specific (PS)/ Genetically modified (GM) |
References |
|---|---|---|---|
| Microcephaly | WRD62 | GM | (15) |
| CDK5RAP2 | PS | (8) | |
| CHMP1A | GM | (16) | |
| Lissencephaly | KATNB1 | GM | (17) |
| Miller Dieker Syndrome | 17p13.3 deletion | PS | (18) |
| Macrocephaly | PTEN | GM | (19) |
| (Seckel syndrome) | CPAP | PS | (20) |
| Periventricular heteropia | PLEKHG6 | GM | (21) |
| DCHS1 and FAT4 | PS & GM | (22) | |
| Schizophrenia | DISC1 | GM | (23) |
| 16p13.11 microduplication | PS | (24) | |
| FGFR1 | PS | (25) | |
| Autism spectrum disorder | CHD8 | GM | (26) |
| Down syndrome | Trisomy 21 | PS | (27) |
| Rett syndrome | MeCP2 | PS | (28) |
| GM1 gangliosidosis | GLB1 | GM | (29) |
| GM2 gangliosidosis/Sandhoff disease | HEXB | PS | (30) |
| Other neural disorders modeled | |||
| Alzheimer’s disease | PSN1 | PS | (31) |
| APOE4 | GM | (32, 33) | |
| Frontotemporal dementia | MAPT | PS | (34) |
| Creutzfeldt-Jakob disease | Exposure to CJD prion subtypes | PS | (35) |
| Hereditary spastic paraplegia | SPG11 | PS | (36) |
| Huntington’s disease | HTT | PS | (37) |
![]()
Go to : 
Region Specific Brain Organoids
Following the early efforts of the Sasai’s group to obtain brain organoids using extrinsic signals, a variety of guided protocols have been established to obtain region specific brain organoids, which have enrichment of cell types specific for the given brain regions with minimal presence of non-relevant cells. The first step in almost all of the guided differentiation protocols is composed of dual SMAD inhibition by administering BMP and TGF-beta receptor inhibitors that suppress the all regulatory-SMAD proteins (38). This inhibition directs the PSC differentiation towards a neuroectodermal lineage and results in the formation of neural epithelium (NE) containing neural progenitor cells (NPCs). These NPCs are further guided towards different regions of the brain by extrinsic signals to obtain region-specific brain organoids (Fig. 1).
Forebrain organoids
During the fetal development, two rostral primary vesicles, telencephalon and diencephalon, develop and form forebrain in adult. Brain organoids that represent these vesicles can be classified into telencephalic organoids and diencephalic organoids. The telencephalic organoids include the cortical organoids (resembling cerebral cortex) and the hippocampal organoids. The diencephalic organoids that have been derived so far represent those for the thalamus, the hypothalamus and the pituitary gland.
Dorsal forebrain/Pallium/Cortical organoids: Cortical organoids are obtained by WNT inhibition that drives NPCs to attain an anterior neuroectodermal fate, to form an anterior neuroepithelium (NE). When cultured in suspension in neural growth medium, the cortical neuroepithelium grow in 3D up to several millimeter in size. Many studies have used this patterning method with some modifications to obtain cortical organoids with high consistency and reproducibility (39-44). The cortical organoids thus obtained displayed the laminar structure with upper and deeper layer cortical neurons, inner and outer radial glia (iRG and oRG), intermediate progenitors, glutamergic neurons, astrocytes, and oligodendrocytes. Impor-tantly, cells in cortical organoids follow similar developmental trajectories to their in vivo counterparts giving rise to active neuronal networks (42, 45-48). As most of the NDDs result from defects in the development of the cerebral cortex, cortical organoids are of prime importance to model NDDs (Table 2).
Table 2
NDDs and other neural disorders modeled so far/can be potentially modeled using region-specific brain organoids
| Region-specific organoid type | Neurodevelopment disorder modelled/Potential application | Modeled gene mutation/environmental condition |
Patient specific (PS)/ Genetically modified (GM) |
References |
|---|---|---|---|---|
| Cortical organoids | Microcephaly | ASPM | PS | (68) |
| Zika virus infection | - | (42, 71) | ||
| Microcephaly associated with Aicardi-Goutières syndrome | TREX1 | PS & GM | (72) | |
| Lissencephaly | LIS1 | GM | (75) | |
| Miller-Dieker Syndrome | 17p13.3 deletion | PS | (76) | |
| Rett syndrome | MeCP2 | GM | (80) | |
| Schizophrenia | DISC1 | PS | (46, 82) | |
| Autism spectrum disorder (ASD) | Idiopathic | PS | (40) | |
| Pelizaeus-Merzbacher disease | PLP1 | PS | (86) | |
| Tuberous sclerosis complex | TSC1 TSC2 | GM | (88) | |
| Japanese encephalitis | JEV infection | - | (89) | |
| MGE organoids | Rett syndrome | MeCP2 | GM | (80) |
| Thalamus organoids | Potential to model epilepsy, ASD, schizophrenia and depression | |||
|
Hypothalamus organoids |
Prader-Willi syndrome | 15q11.2-q13 deletion | PS | (55) |
| Pituitary organoids | Congenital pituitary hypoplasia | OTX2 | PS | (95) |
| Cerebellum organoids | Medulloblastoma | SMARCA4 | Overexpression of mutant protein | (96) |
| Potential to model spinocerebellar ataxia (SCA), Dandy-Walker syndrome and Angelman syndrome | ||||
| Assembloids | Timothy syndrome (assembly of pallial and subpallial organoids) | CACNA1C | PS | (45) |
| Potential to model neuropsychiatric disorders | ||||
| Other neural disorders | ||||
| Cortical organoid | Alzheimer’s disease |
APP duplication PSEN1 |
PS | (101) |
| PSEN1 | PS | (102) | ||
| Hippocampus | Potential to model cognitive dysfunctions due to Alzheimer’s disease | |||
| Midbrain | Parkinson’s Disease | LRRK2 | GM | (103, 104) |
![]()
Ventral forebrain/Subpallium/MGE organoids: The medial ganglionic eminence (MGE) organoids or subpallium organoids are obtained by WNT inhibition along with SHH activation to direct the anterior NE towards a ventral fate. The resulting MGE organoid progenitors are validated by the presence of ventral forebrain (subpallial) marker NKX-2.1 accompanied by high levels of FOXG1 and the absence of dorsal forebrain (pallial) marker EMX1. Mature MGE organoids contain primarily inhibitory GABAergic neurons as opposed to excitatory glutamergic neurons of the cortical organoids (44, 45, 49).
Hippocampal organoids: The hippocampal organoids are generated by directing the anterior NE towards the dorso-medial forebrain fate by extrinsic activation of WNT and BMP signaling for a specific time window. The organoids obtained by this method resemble a developing hippocampus tissue that on a long term culture gives rise to hippocampus specific neurons like pyramidal neurons, hippocampal granule neurons as well as astrocytes (50, 51). Another report from Pomeshchik et al. combined SHH inhibition with WNT activation to specify cells toward a dorsal telencephalic fate. Further culturing of these organoids with brain-derived neurotrophic factor (BDNF) allow expansion of hippocampal neural progenitors leading to cellular and structural signatures similar to hippocampus (52).
Thalamic organoids: Caudalization or posterior neuroectoderm specification is a crucial step in obtaining diencephalic forebrain organoids. To obtain thalamic organoids Xiang et al. (53) induced caudalization in the NE by avoiding WNT inhibition and by administering a known caudalizing factor, insulin during dual SMAD inhibition. In addition, to prevent excessive caudalization, a mitogen-activated protein kinase-extracellular signal regulated kinase (MEK-ERK) inhibitor was used at a later time point. The organoids were further supplemented with human BMP7 to promote thalamic differentiation (53). These organoids contained cells that expressed thalamus specific markers significantly higher than the cortical organoids and lacked enriched cortical neurons ensuring their developing thalamus like identity.
Hypothalamus organoids: To obtain hypothalamus organoids, the NE is directed towards the ventral-rostral lineage mediated by WNT and SHH signaling for a specific time window. After this, the pre-hypothalamic organoids are allowed to mature in a spin reactor with FGF-2 and CTNF supplementation. These organoids contained cells that expressed early and late hypothalamus markers in the developing and mature stage respectively (42). Another independent group has showed that the neuroepithelium attains the ventral-rostral lineage only with SHH signaling and strict absence of other signaling factors (54). Recently, Huang et al. established a protocol to obtain a sub-region of hypothalamus, the arcuate nucleus, using combined SHH signaling activation and WNT signaling inhibition. The resulting organoids exhibited the cell type diversity and molecular signatures similar to the arcuate nucleus of hypothalamus (55).
Pituitary organoids: Unlike other forebrain regions, the pituitary arises from the oral ectoderm and not the neuroectoderm. The development of pituitary is dependent on its contact with adjacent hypothalamus. To obtain pituitary organoids, Ozone et al. (54) modified their hypothalamus derivation protocol by exposing the NE only to SHH and BMP signaling and strictly avoiding other signaling. By this method they demonstrated the co-induction of both the oral ectoderm and the ventro-rostral NE. The ventro-rostral NE matured into hypothalamus like tissue and was able to induce the oral ectoderm to invaginate and develop into Rathke’s pouch (pituitary primordium) like structures. The pituitary progenitors in these organoids differentiated into mature corticotroph and somatotroph-like cells that secreted adrenocorticotropic hormone (ACTH) and growth hormone (GH), respectively (54). This work demonstrated that the hypothalamic-pituitary interaction during the development process is crucial for pituitary organogenesis and can be recapitulated in vitro.
Choroid Plexus organoids: Choroid plexus is a secretory epithelial tissue lining each of the four ventricles in the central nervous system. Its main function is to produce cerebrospinal fluid (CSF) but it also creates a barrier that regulates entry of nutrients and small compounds into the brain. Cerebral organoids predominantly contain structures with forebrain identities that also include choroid plexus epithelium. To enrich choroid plexus identity, Pelegrini et al. (56) developed a protocol based on unguided cerebral organoid generation with the addition of dorsalizing factor Bmp4 and the Wnt signaling activator CHIR. The resulting organoids exhibited enrichment of choroid plexus tissue that produces CSF and forms in vitro barrier with a similar selectivity to small molecules as in vivo choroid plexus tissue (56).
Midbrain organoids
Midbrain organoids are derived by mimicking the signal cues experienced at the center of rostral-caudal axis in the developing brain. This is achieved by administering both SHH and WNT3A agonists to the developing NE to attain a midbrain floor plate-like identity and further maintaining them in neurotrophic factors like brain derived neurotrophic factor (BDNF) and glial derived neurotrophic factor (GDNF). The resulting midbrain organoids contained post-mitotic midbrain dopaminergic neurons (mDANs) validating the midbrain-like identity of the organoids (42, 57, 58). A recent report by Nickels et al. (59) demonstrated the derivation of more reproducible and viable midbrain organoids by adjusting the extrinsic signaling timeline and initial cell count.
Hindbrain organoids
The hindbrain organoids can be classified into metencephalic organoids and myelencephalic organoids. Cere-bellar organoids are the metencephalic organoids that are characterized by the presence of Purkinje cells. In an attempt to derive cerebellar organoids with Purkinje cells (60), initiated NE commitment in PS cells by TGF beta inhibition and caudalization by administering FGF2 and insulin. The resulting cerebellar NE expressed FGF8 and WNT1, which are implicated in the function of isthmic organizer that marks the midbrain-hindbrain boundary (MHB). The cerebellar NE was allowed to mature with intermittent FGF19 and SDF1 (stromal derived factor-1) treatment, to promote the formation of a continuous cerebellar NE with dorsal-ventral polarity and for the generation of different cerebellar progenitors – Purkinje cells, Golgi cells, granular cells, DCN (deep cerebellar nuclei) progenitor neurons along with non-Golgi type inter-neurons. A recent report, although not by guided protocol, showed an attempt to derive brainstem which consists of the midbrain, pons (metencephalon) and medulla oblongata (myelencephalon) (61).
Brain assembloids
Brain assembloids include 1) fused region-specific brain organoids, 2) region-specific brain organoids co-cultured with other non-neuronal but physiologically relevant cells/tissues and finally 3) brain organoids co-cultured with organizers that releases a gradient of extrinsic signaling to mimic the dorsal-ventral and anterior-posterior axis determinants of the fetal brain. Many studies have fused relevant region-specific brain organoids to study interneuron migration (44, 45, 49, 62), neuronal projections and oligodendrogenesis (53). Methods to create region-specific brain organoids with vasculature for their better survival have been reported (63-65). In addition, region-specific brain organoids have also been created with integrated microglia that can be used to study microglial migration and response to injury in a 3D environment (66). Fairly recently, Cederquist et al. (67) demonstrated an ingenious approach of creating and using SHH gradient releasing organizers to mimic the dorsal-ventral axis in the organoids.
Go to :
NDDs Modeled with Region-Specific Brain Organoids
Region-specific brain organoids are used increasingly to model different NDDs due to their higher relevance to the respective NDD modeled. A complete list of NDDs that have been modeled using region-specific brain organoids so far are shown in Table 2.
Microcephaly
Microcephaly in humans is a condition with abnormal growth of the brain during embryonic development or in the first year after birth, resulting in decreased brain size (especially the cerebral cortex) and a smaller head. Micro-cephaly can be a result of a single mutation (primary microcephaly), viral infection during pregnancy (e.g., ZIKV induced microcephaly) or could be a manifestation of other syndromes (Seckel syndrome, Aicardi-Goutières syndrome). Cortical expansion and folding is a key process in human brain development. Cortical organoids recapitu-late the expansion and laminized organization of the layered neocortex generated from the radial glial (RG) cells and intermediate progenitor (IP) cells (68), making them an ideal model system to study microcephaly.
Autosomal recessive primary microcephaly (MCPH) in humans, is a neurogenic mitotic disorder with deficient neurogenesis, whereas other neuronal functions are preserved. Twelve genetic loci (MCPH1-12) with mutations have been mapped to be associated with MCPH, making it an heterogenous disorder. Based on the genes mapped, it is predicted that the MCPH disease phenotype may be due to a disturbed mitotic spindle orientation, premature chromosomal condensation, signaling response as a result of damaged DNA, microtubule dynamics, transcriptional control or a few other hidden centrosomal mechanisms that can regulate the amount of neuronal population that arise from NPCs (69). ASPM (abnormal spindle-like microcephaly-associated protein) related primary microcephaly (MCPH5) has been modeled using cortical organoids (68). The ASPM mutant cortical organoids exhibited early cortical development defect characterized by reduced tissue size, reduced NPCs, and a defective lamination with fewer RG cells and reduced neuronal activity. This study reveals that dysfunction of ASPM could result in defective neurogenesis and loss of cortical lamination leading to defective neuronal activity in MCPH5 patients.
Zika virus (ZIKV) has been shown to infect the developing fetal brain and cause microcephaly (70). Many studies have modeled ZIKV mediated microcephaly in cerebral organoids (Table 1). Qian et al. (42), performed an in-depth analysis of cortical organoids that were transiently exposed to ZIKV at different developmental stages. They show that ZIKV exhibits tropism towards NPCs over intermediate neural progenitors or immature neurons, resulting in smaller cortical organoids. They showed that infected NPCs produced more infectious viral particles leading to an increasing number of infected cells over time (42). The reduction in infected organoid size was not only due to reduced NPC proliferation but also due to increased cell death of NPCs and other uninfected neurons. Further they also add that ZIKV infection-induced NPC death was much more dramatic in cortical organoids than seen in monolayer cultures. In summary, using cortical organoids, this work identified common mechanisms between ZIKV induced and primary microcephaly i.e. loss of NPCs. The same research group in their follow up study, systematically introduced individual proteins encoded by ZIKV and found that ZIKV-NS2A reduced radial glial cell proliferation and caused adherens junction complex deficits in cortical organoids, thus revealing the pathogenic mechanism underlying ZIKV infection during fetal brain development (71).
Aicardi-Goutières syndrome associated microcephaly: Aiacrdi-Goutières syndrome (AGS) is a neuroinflammatory disorder with severe and persistent intellectual and physical problems, with an onset in early infancy. Mutations in antiviral genes such as TREX1 has been known to cause AGS. TREX1 codes for an enzyme known as ‘three-prime repair exonuclease I’ (TREX1). Loss of TREX1 has been predicted to result in the accumulation of endogenous reverse-transcribed L1 DNA species leading to neuroinflammation and neurotoxicity leading to microcephaly. Microcephaly associated with AGS has been modeled using cortical organoids lacking TREX1 (72). TREX1-deficient organoids displayed a significant reduction in diameter during the later stages of differentiation due to neuronal cell death. In addition, treatment with reverse transcription inhibitors rescued both size of the organoids and toxicity mediated cell death, emphasizing potential therapeutic strategies to treat patients suffering from AGS (72).
Lissencephaly
Cortical folding during its expansion is crucial in brain development enabling the efficient packaging of billions of neurons in the limited cranial space. Reduced cortical folding resulting in smooth brains with abnormal/absent gyrification is one of the manifestations of a severe neurodevelopmental disorder known as Lissencephaly, which is accompanied by intellectual disability and reduced life expectancy. Mutation in several genes have been reported to be associated with lissencephaly including LIS1, DCX and TUBA1A that are involved in formation and regulation of microtubules, which play an important role in the developing brain (73). LIS1 protein functions as a component of an intracellular multiprotein complex including NDEL1 and 14.3.3ε, is involved in several key functions including proliferation, neuronal migration, regulation of molecular motors and cytoskeleton (74). LIS1 mutation is the most common cause of isolated lissencephaly in humans. To study the effect of LIS1 mutation in brain development, Karzburn et al. (75) established methods in cortical organoids to study the biophysical and biomechanical mechanisms exhibited by fetal brain during cortical folding. Using these physical quantification methods they compared control organoids with organoids generated from isogenic LIS1 mutant lines and found that the mutant organoids displayed reduced wrinkling with changes in their nuclear motion. In addition, mutant organoids displayed a significant differential regulation of the ECM and cytoskeleton genes compared to the control organoids, signifying the role of LIS1 in nuclear cytoarchitecture during brain development (75).
LIS1 resides close to YWHAE (codes for 14.3.3ε) at the 17p13.3 locus in the human genome. The loss of this 17p13.3 locus results in Miller-Dieker syndrome (MDS) which exhibits severe lissencephaly phenotype due to increased structural alterations in the cortical architecture compared to isolated lissencephaly. The MDS organoids derived by Iefremova et al. (76) from MDS patients with 17p13.3 chromosomal deletion, expanded slowly when compared to control organoids. The observed reduction in the expansion rate was found to be associated with a switch in the division mode of ventricular radial glia (vRG) cells from symmetric to asymmetric cell division. Further, MDS organoids displayed altered microtubule network demonstrated by irregular lining and distribution of N-cadherin at the apical surface of the rosettes. Interestingly, this altered microtubule network and disruption of cortical niche architecture resulted in a non-cell-autonomous disturbance of the N-cadherin/β-catenin signaling axis. Most importantly, pharmacological activation of Wnt signaling in MDS organoids rescued the disease phenotype and ameliorated the growth defects (76).
Rett syndrome
Rett syndrome (RTT) is a severe X-linked neurodeve-lopmental disorder without effective therapies. Being an X-linked disorder, RTT is rarely observed in males, however leading to severe neonatal encephalopathy and early lethality. In females, the disease symptoms appear 6 months after normal development post birth. The symptoms include deceleration of head growth, gait abnormalities, breathing disturbances and loss of spoken language and hand use with the development of distinctive hand stereotypies (77). Majority of the cases of RTT are caused by mutations in MeCP2 coding for methyl-CpG binding protein 2 (78). MeCP2 has a broad role in maintaining the genome-wide transcription homeostasis by involving in suppression/activation of transcription, chromatin remodeling, alternative splicing and microRNA processing (79). In their attempt to model RTT in vitro (80), derived human MGE organoids and human cortical organoids from PS cells modified to carry mutations in MeCP2. Interestingly mutant organoids exhibited severe abnormalities specific to interneurons (IN) but not cortical neurons. They found that the abnormality in INs was mainly due to the genome wide binding of bromodomain-containing protein 4 (BRD4), a co-regulatory protein involved in gene activation. The persistent hyperactive transcription mediated by BRD4 binding is detrimental to neural development, resulting in impaired neuronal activity and plasticity. Finally, they demonstrated that low dose of JQ1 (cancer drug that inhibits BRD family proteins) treatment could impede chromatin binding of BRD4, recover genome architecture and transcription and consequently reverse the development defect of Rett syndrome observed in MeCP2 mutant organoids (80). All put together, this work showed that BRD4 dysregulation is a critical driver for RTT etiology and suggest that targeting BRD4 could be a potential therapeutic opportunity for RTT.
Schizophrenia
Schizophrenia is a serious mental disorder involving a range of problems with thinking (cognition), behavior and emotions. Schizophrenic patients interpret reality abnormally leading to delusions, hallucinations and abnormal thinking. DISC1 (disrupted-in schizophrenia 1) which is known to be associated with major mental illnesses, is also known to be associated with schizophrenia. It is believed that DISC1 acts as an intracellular anchoring protein interacting with hundreds of proteins involving in cortical development (81). In order to better understand pathogenic effects of DISC1 mutation in schizophrenia, Ye et al. (82) developed cortical organoids from patients with DISC1 lacking the C’ terminal region (83) that is essential for its binding to an interactive protein complex called Ndel1/Nde1 (84). Due to the defect in interaction between DISC1 and Ndel1/Nde1, the cortical organoids displayed radial glial cells with delayed cell cycle progression, a phenomenon observed in 2D cell culture and in animal models (82). The same research group in their further studies had developed sliced cortical organoids from a patient with psychiatric disorder (carrying a frameshift mutation in DISC1) and showed that mutant sliced cortical organoids exhibited deficits in cortical neuron subtype differentiation and failed to form segregated upper and deep cortical neurons (46).
Autism spectrum disorder (ASD)
Autism is known as a “spectrum” disorder owing to the wide variation in the type and severity of symptoms people experience. With that being said, the most commonly observed symptoms of ASD are language deficits, social communication difficulties and repetitive behaviors (85). Rare gene mutation identified to be associated with ASD account for 1∼2% of the ASD cases, leaving a large number of cases with no clear etiology. Mariani et al. (40) developed ASD specific cortical organoid model from iPSCs derived from idiopathic ASD patients with macrocephaly. Genome wide transcriptomic analysis of ASD cortical organoids showed a gene expression profile of altered neurodevelopment i.e. increased cell proliferation, unbalanced excitatory/inhibitory neuron differentiation and increased synaptic development with an increase in expression of FOXG1 (involved in telencephalon development). The increase in synapse formation was further confirmed by cytologic analysis and electrophysiology experiments. The altered neurodevelopment in these ASD organoids were attributed to increased FOXG1 expression and RNA interference of FOXG1 resulted in a balance in the development of excitatory and inhibitory neurons in ASD organoids (40). This work demonstrates the immense use of cortical organoids to understand the disease pathology in idiopathic conditions, offering drug targets.
Pelizaues-Merzbacher disease
Pelizaues-Merzbacher disease (PMD) is an X-linked leukodystrophic condition involving the central nervous system (the brain and the spinal cord). Leukodystrophies are conditions that affect the myelinated nerve cells that form the white matter (hence the name Leukodystrophy). PLP1 which is involved in myelination is known to be associated with this disease. Madhavan et al. (86) developed protocols to obtain cortical organoids containing myelinating oligodendrocytes expressing PLP1 (phospholipid protein 1), the most abundant oligodendrocyte membrane lipid protein and MYRF, a transcription factor specifically expressed in the oligodendrocytes of the CNS. Continuing with that, they showed that oligocortical organoids from PMD patients with defective PLP1 contained oligodendrocytes but the membrane protein PLP1 was accumulated in the perinuclear space. This accumulation resulted in ER stress and cell death in oligodendrocytes explaining the loss of myelination in PMD patients. Finally they demonstrate that subsequent treatment of PLP1 mutant cortical organoids with protein-kinase-R-like ER kinase (PERK) inhibitor, improved correct mobilization of PLP1 into oligodendrocyte processes (86).
Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is a multisystem developmental disorder characterized by multiple benign hamartomas in various organs including brain with a wide range of clinical features like seizures, autism, intellectual disability, facial angiofibromas, cardiac rhabomyomas including the hallmark pathology of the presence of cortical tubers. Cortical tubers are regions of dysmorphic and disorganized neurons and glia in the cortex (87). TSC manifests as a result of compound heterozygous mutations in TSC1 and TSC2, coding for hamartin and tuberin respectively, that are negative regulators of mTORC1 (mechanistic target of rapamycin complex 1) signaling. Using cortical organoids that carried a heterozygous TSC2 mutation and introduction of a second mutation in the functional allele during neural progenitor expansion, Blair et al. (88) showed that the second-hit during neural progenitor expansion is required for the formation of cortical tubers. Finally by blocking mTORC1 signaling using rapamycin at different time points, they demonstrate that early rapamycin treatment significantly reduced the formation of cortical tubers and late rapamycin treatment has no effect (88). Human cortical organoids are extremely useful in these kinds of disease modeling as cortical tuber formation is specific to humans and do not develop in TSC mouse models.
Japanese encephalitis
Japanese encephalitis is caused by a mosquito-borne flavivirus known as JEV (Japanese encephalitis virus). Although most of the JEV infection is asymptomatic, the case fatality ratio of the affected population is 20∼30%. Among survivors, 30%∼50% have serious neurologic, cognitive, or psychiatric dysfunctions. JEV exposed to human cortical organoids preferentially infected astrocytes and the progenitors of outer radial glial cells, leading to reduced proliferation and cell death (89). Infecting the organoids at various stages of development revealed that interferon signaling pathway activation in response to JEV infection occurred only in older organoids (older than 8 weeks) but not the younger organoids (4 weeks old or lesser), explaining the more severe outcomes of JEV infection in younger population (89).
Congenital pituitary hypoplasia
Congenital pituitary hypoplasia (CPH) is a genetic condition with impaired pituitary development leading to hypopituitarism. Mutations in around 9 genes encoding transcription factors including OTX2 are known to be associated with CPH (90). As OTX2 plays a significant role in the development of forebrain, eye and the pituitary, mutation in OTX2 have been observed to cause impaired forebrain development, malformation of the eye in addition to impaired pituitary development (91-93). Earlier reports have shown that OTX2 expression in hypothalamus is required for anterior pituitary development (94). To understand the pathogenic effect of OTX2 mutation in pituitary development (95) derived CPH patient specific organoids containing both oral ectodermal tissues (that develops into pituitary) and hypothalamic tissues. Unlike control organoids, the oral ectoderm of the mutant organoids could not differentiate into ACTH and GH hormone producing cells displaying impaired pituitary development. Finally with their chimeric organoid culture approach they show that OTX2 from the hypothalamus is required for pituitary development through a OTX2-FGF10-LHX3 pathway (95), thus unraveling the molecular mechanism behind pituitary development and CPH pathogenesis.
Medulloblastoma
Medulloblastoma (MB) is the most common malignant brain tumor in children that initiates at the cerebellum. Of all the known oncosuppressors, mutation in SMARCA4 is most frequently associated with Group 3 MB (severe form of medulloblastoma). In an attempt to create a Group 3 MB organoid model, Ballabio et al. (96) identified ‘Otx2 and c-MYC’ (OM) as MB driver genes and overexpressed them through electroporation. The resulting OM organoids contained cerebellar progenitors that were over-proliferating and with impaired differentiation. Additionally SMARCA4 overexpression in OM organoids reduced the proliferation of cerebellar progenitors, conveying that SMARCA4 might have a role in reducing OM-induced Group 3 MB. Co-overexpression of SMARCA4 T910M (protein product of most common SMARCA4 mutation) along with SMARCA4 WT, they found the mutant protein has the ability to block the oncosuppressing activity of the WT protein. Together, this study revealed that SMARCA4 T910M represses SMARCA4 WT functions and act as a dominant-negative in Group 3 MB patients (96).
Timothy syndrome
Timothy syndrome (TS) is a multisystem developmental disorder that primarily affects the heart but can affect many other areas of the body, including the nervous system. Clinical features of TS are cognitive impairment and autism, congenital heart defects, syndactyly and immune deficiency. Mutation in CACNA1C, coding for CaV1.2 is widely associated with this disease (97). CaV1.2 is a voltage-dependent L-type calcium channel protein (LTCC) that regulates the intracellular calcium levels in the excitable cells of heart and brain. By their assembloid modeling approach, Birey et al. (45) fused cortical (pallium) organoids and sub-pallium organoids derived from TS patients with CACNA1C mutation and showed interneuron migration between organoids with increased neuronal saltation frequency but decreased length and speed, suggesting the presence of abnormal cortical development and function in TS. They finally showed that application of LTCC blockers can rescue the migration defects exhibited by the interneurons between organoids.
Prader-willi syndrome
Prader-Willi syndrome (PWS) is a genetic-based neurodevelopmental disorder caused by the absence of paternally expressed genes located in the 15q11.2-q13 chromosome region (98). In newborns, symptoms associated with PWS include diminished muscle tone (hypotonia), feeding difficulties, impaired satiety, obesity as well as social and learning deficits (99). Impaired development of hypothalamus and eventual dysregulation of its function is the cause of several symptoms of PWS particularly due to endocrine dysfunctions (100). Recently, Huang et al. (55) generated hypothalamic arcuate organoids (ARCOs) from healthy and PWS patient iPSCs for modeling and identifying cellular and molecular neurodevelopmental abnormalities associated with PWS. ARCOs derived from patient iPSCs were significantly larger in size compared to control ARCOs. This was due to an increased percentage of proliferating neural progenitors and a decrease percentage of mature neurons suggesting neuronal differentiation defects in the arcuate nucleus during the early progression of PWS (55). One cellular hallmarks of the arcuate nucleus is the release of melanocyte-stimulating hormone (MSH) in response to leptin. ARCOs treated with leptin showed a substantial increase in MSH level in control, although this increase was miniscule in PWS patient-derived ARCOs (55). Together, this study revealed developmental and functional deficits of the arcuate nucleus in the hypothalamus associated with the pathophysiology of PWS.
Go to :
Potential NDDs that Can Be Modeled Using Region-Specific Brain Organoids
Although human forebrain organoids have been extensively used to model NDDs and other neural disorders, there is still a mountain of disorders that can be studied with region-specific brain organoids. For example, thalamic organoid disease models will be excellent model systems to study epilepsy, ASD, schizophrenia and depression. Disorders like spinocerebellar ataxia (SCA), Dandy-Walker syndrome and Angelman syndrome can be better studied with cerebellum organoid disease models.
Go to :
Other Neural Disorders Modeled Using Region-Specific Brain Organoids
Molecular and structural alterations during brain development are believed to manifest as neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Frontotemporal dementia (FTD) in the later phase of life. With respect to that, region-specific cortical organoids have also been used in modeling AD and PD. Cortical organoids from AD patients recapitulated AD-like pathologies such as amyloid aggregation, hyperphosphorylated tau protein and endosome abnormalities in an age dependent manner (101). Treatment of AD cortical organoids with β- and γ-secretase inhibitors significantly reduces amyloid and tau pathology showing the potential of this model system in drug discovery for AD (101). Another study showed a significantly higher level of Aβ42 (main component of amyloid aggregation), hyperphosphorylated tau protein, increased expression of proinflammatory cytokines, alterations in ECM composition followed by cell death, mimicking the AD-related cytotoxicity (102). With respect to PD, two independent studies have derived PD midbrain organoids and demonstrated that the mutant midbrain organoids exhibit neurodevelopment defect marked by increased aggreagion of α-synuclein and its aberrant clearance and decrease in the number and complexity of midbrain dopaminergic neurons (mDANS) recapitulating the three dimensional pathological hallmarks observed in PD (103, 104).
Go to :
Conclusions
Human neurodevelopmental disorders are difficult to study because of the limited accessibility to the developing human fetal brain. Animal models can be useful to an extent but species barrier remains an issue. Especially human cerebral cortex is much evolved when compared to other vertebrates including non-human primates. Also, a number of drugs that have shown promising results in animal models have failed in human clinical trials. In addition, diseases associated with variations/mutations in the non-coding regions of the genome are not well conserved between species. Human brain organoids addresses all these concerns. Region specific brain organoids are reproducible with uniformity, consistency across various PS lines. It is one’s choice to prefer cerebral organoids or region-specific brain organoids, depending on the scientific question one wants to address. Assembly of complex brain regions with different region-specific brain organoids with modified protocols and bioengineering techniques would ideally result in next generation brain organoids, that would help in better understanding of NDD disease mechanism and its treatment.
Go to :
 XML Download
XML Download