

 PDF
PDF Citation
Citation Print
Print


Introduction
Embryonic stem (ES) cells are derived from in vitro-fertilized embryos and are established using the inner cell mass of blastocysts (1). ES cells can self-renew, maintaining their stemness, and have pluripotent ability of differentiation to multiple lineages under certain conditions. Understanding of the molecular mechanisms involved in the maintenance of ES cells is one of the most critical issues in stem cell biology.
Reactive oxygen species (ROS) are defined as chemically reactive chemical species containing oxygen. A certain amount of ROS is generated during cellular metabolism and has an important role in cell signaling and homeostasis (2). However, excessive ROS causes oxidative stress and induces cell damage and cell death (3). ES cells are highly proliferative cells that require robust metabolism to enhance rapid division. However, this process relies primarily on glycolysis and the pentose phosphate pathway rather than on oxidative phosphorylation (4-6). Furthermore, ES cells have higher expression of antioxidant genes than differentiated cells (7, 8). Therefore, ES cells have low levels of ROS, which is essential to maintain pluripotency.
MUC1 is a transmembrane protein that is found in various cells and controls adhesion, migration, and cell signaling (9). MUC1 is composed of an extracellular subunit and a membrane–associated subunit. The N-terminal subunit of MUC1 (MUC1-N) is heavily glycosylated and comprises most of the variable number tandem repeat region with a signal peptide sequence at the N-terminus and the sea urchin sperm protein enterokinase and agrin (SEA) domain. The SEA domain includes an autoproteolytic cleavage site, which generates MUC1-N and MUC1-C terminal (MUC1-C) subunits soon after its translation as a single protein. MUC1-C comprises an extracellular domain (58 amino acid), a transmembrane domain (28 amino acid), and a cytoplasmic tail (72 amino acid) (10). Under normal conditions, MUC1 subunits form a heterodimeric complex on the plasma membrane by association via hydrogen bonds. However, the complex can be dissociated by additional proteolytic cleavage events and released from the cell surface.
MUC1-C interacts with various signaling molecules such as STAT3 (11), GSK3β (12), and NF-κB (13) and participates in their downstream signaling pathways. Furthermore, MUC1-C associates with various transcription factors and transcriptionally regulate expression of their target genes on the promoters (10). These signaling molecules are involved in the regulation of pluripotency in ES cells (14-16). However, the role of the MUC1-C regarding ES cells remains unknown. Therefore, this study aimed to check the effects of MUC1-C inhibition and draw the possible function of MUC1-C in mouse ES (mES) cells.
Go to : 
Materials and Methods
Mouse embryonic stem cell culture
The mES cell line R1 was maintained on mouse embryonic fibroblast (MEF) cells in ES medium containing Dulbecco’s modified Eagle’s medium (DMEM), 15% fetal bovine serum (FBS; Hyclone Inc., Logan, UT, USA), 0.1 mM β-mercaptoethanol, 0.1 mM nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco/Life Technologies, Long Island, NY, USA) at 37℃ under a humidified atmosphere of 5% CO2. Cells were dissociated with TrypLE Express (Gibco) and then seeded on the MEF cells prepared as follows. MEF cells were harvested, irradiated with 50 Gray, and seeded at a density of ∼5.5×104 cells/ml in MEF medium (DMEM, 10% FBS, 0.1 mM β-mercaptoethanol, 0.1 mM nonessential amino acids) a day before mES cell seeding.
Differentiation of mES cells with retinoic acid
The cells were plated on gelatin-coated six-well plates and grown overnight with Leukemia Inhibitory Factor (LIF; Peprotech, Rocky Hill, NJ, USA). To stimulate the differentiation of mES cells, cells in the MEF medium were treated with 10 μM retinoic acid (RA; Sigma Aldrich, St. Louis, MO, USA) for 7 days. The medium was changed every day.
Flow cytometry analysis
The expression of MUC1 was assessed by flow cytometry. The cells were fixed and permeabilized using cytofix/cytopermTM (BD Biosciences, San Jose, CA, USA). After washing, the cytofix/cytopermTM wash solution (BD Biosciences) was added to the cells together with anti-MUC1 antibody (Abcam, Cambridge, MA, USA) recognizing the C-terminal domain of MUC1 and incubated at 4℃ overnight. The cells were washed with the cytofix/cytopermTM wash solution and then incubated with the Alexa 488-labeled secondary antibody (Invitrogen, Carlsbad, CA, USA) in cytofix/cytopermTM wash solution at 4℃ in the dark for 1 hr. Samples were analyzed by a FACSCalibur (BD Biosciences) flow cytometer and the data were analyzed with the software FCS Express 5 (De Novo Software, Pasadena, CA, USA).
Treatment with MUC1-C inhibitor peptide
The cells were plated on gelatin-coated six-well plates and grown overnight with LIF. Cells were treated with MUC1-C inhibitor GO201 (D-RRRRRRRRR-CQCRRKN YGQLDIFP) or control peptide CP1 (D-RRRRRRRRR-AQARRKNYGQLDIFP) at the indicated concentrations for 24 hr. CP1 and GO201 peptides were synthesized by Peptron Inc. (Yuseong-gu, Daejeon, Republic of Korea) as described previously (17).
RT-PCR and quantitative PCR
Total RNA was isolated using TRI ReagentⓇ according to the manufacturer’s instructions (MRC, Cincinnati, OH, USA). 2 μg of total RNA was reverse-transcribed in the first-strand buffer containing 6 μg/ml oligo(dT) primer, 50 U M-MLV reverse transcriptase (Invitrogen), 2 mM dNTP, and 40 U RNaseOUTTM recombinant ribonuclease inhibitor (Invitrogen) at 42℃ for 1 hr. Quantitative PCR reaction was performed using Rotor-Gene SYBRⓇ Green PCR Kit (Qiagen, Germany). The PCR reaction was performed at 10 min at 95℃ for 1 cycle, 15 sec at 95℃ and 20 sec at 60℃ for 40 cycles. The primer sequences used are as follows: qOct4, 5’-CACGAGTGGAAAGCAACTC A-3’ (sense) and 5’-AGATGGTGGTCTGGCTGAAC-3’ (antisense); qMUC1, 5’-GGTTGCTTTGGCTATCGTCTA TTT-3’ (sense) and 5’-AAAGATGTCCAGCTGCCCATA-3’ (antisense); qGAPDH, 5’-AACTTTGGCATTGTGGAAGG-3’ (sense) and 5’-ACACATTGGGGGTAGGAACA-3’ (anti-sen-se). Relative expression of the target gene was calculated using the ΔΔCt method with GAPDH as a reference control. Relative expression of the genes during differentiation (%) was determined by dividing normalized values within a data set by the normalized arbitrary units of undifferentiated mES cells. A standard PCR reaction was performed using one microliter of the cDNA solution for 20∼30 cycles of denaturation for 60 sec at 95℃, annealing for 60 sec at 58℃, and elongation for 60 sec at 72℃. The primer sequences used are as follows: Oct4, 5’-CTCGA ACCACATCCTTCTCT-3’ (sense) and 5’-GGCGTTCTCTT TGGAAAGGTGTTG-3’ (antisense); MUC1, 5’-GCAGTGTG CCAGTGCCGCCG-3’ (sense) and 5’-CAGTCCTTCTGA GAGCCACC-3’ (antisense); GAPDH, 5’-ACCACAGTCCA TGCCATCAC-3’ (sense) and 5’-TCCACCACCCTGTTG CTGTA-3’ (anti-sense).
Western blotting
Whole cell lysates were prepared as previously described (18). Samples were separated by SDS-polyacrylamide gel electrophoresis and electro-transferred to polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Billerica, MA, USA). Membranes were blocked with 5% dry skim milk and treated with anti-Phospho-Stat3 (Tyr705), anti-Stat3 (Cell Signaling Technology, Beverly, MA, USA), anti-Oct4 (BD Biosciences), anti-MUC1 (Abcam), anti-Bcl2, anti-Mcl1, anti-GAPDH, and anti-β-actin (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) antibodies. Immunoreactive proteins were visualized by horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology Inc.) and an ECL reagent (ATTO Corp., Tokyo, Japan).
Alkaline phosphatase assay
Alkaline phosphatase staining was conducted using alkaline phosphatase kit 86-R (Sigma) according to the manufacturer’s protocol. Briefly, cell layers were rinsed with phosphate buffer saline (PBS) and fixed with citrate-acetone-formaldehyde fixative solution (65 ml acetone, 0.8 ml of 37% formaldehyde, 25 ml citrate solution) at room temperature for 30 sec. The samples were then stained with an alkaline-dye mixture (Sodium Nitrite Solution: FRV-Alkaline Solution: Naphthol AS-BI Al-kaline=1:1:1) at room temperature for 15 min.
Cell proliferation assay
A cell proliferation assay was conducted using Cell Proliferation ELISA, BrdU kit (Roche Diagnostics. GmbH, Mannheim, Germany) according to the manufacturer’s instructions. Briefly, the cells were placed onto a 96-well plate, and incubated for 24 hr. The cells were treated with GO201 or CP1 at the indicated concentration for 24 hr. After treatment, 10 μM BrdU was added to each well and incubated for 4 hr at 37℃. After the incubation, the growth medium was removed and cells were denaturated and fixed using FixDen for 30 min. The fixed cells were then incubated with anti-BrdU antibody conjugated with peroxidase (anti-BrdU-POD) for 90 min. The peroxidase substrate was added and BrdU incorporation was quantified by measuring the absorbance at 450 nm using an ELISA reader.
Cell cycle analysis
The cells were treated with CP1 or GO201 at 2.5 μM for 24 hr, harvested, and fixed in 70% ethanol at 4℃ overnight. The next day the fixed cells were stained with propidium iodide (PI)/RNase staining buffer (BD Bio-sciences) for 15 min and analyzed using a FACSCalibur flow cytometer (BD Biosciences). Cell debris and clumps were excluded from the analysis for all samples. The data were analyzed with the software FCS Express 5 (De Novo Software).
Analysis of apoptosis (Annexin V/PI staining)
To determine the cellular apoptosis or necrosis, we used the Annexin-V-FLUOS staining kit (Roche). The cells were dissociated with TrypLE Express, washed with PBS, and stained with Annexin-V-FLUOS labeling solution for 15 min. The cells were then analyzed by a FACSCalibur (BD Biosciences) flow cytometer.
Reactive oxygen species (ROS) measurement
For the measurement of the intracellular ROS levels, we used an oxidation sensitive fluorescent probe, 2’,7’-dichlorofluorescein diacetate (DCF-DA) (Molecular Probes, Eugene, OR, USA). Briefly, the cells were treated with GO201 or CP1 at the indicated concentrations for 24 hr. As another experiment, the cells were untreated or treated with N-acetyl-L-cysteine (NAC) at the indicated concentration for 1 hr followed by treatment with GO201 or CP1 at the indicated concentrations for 24 hr. After treatment, the cells were incubated with 10 μM DCF-DA for 30 min at 37℃. Then the cells were dissociated with TrypLE Express, suspended in fresh medium, and analyzed by a FACSCalibur (BD Biosciences) flow cytometer.
Determination of glutathione (GSH) levels
For the measurement of the intracellular glutathione levels, we used a Glutathione Colorimetric Assay Kit (BioVision Inc., Milpitas, CA, USA). The cells were harvested with ice-cold PBS and lysed in ice-cold glutathione buffer. The cell lysate was then incubated with glutathione reductase and the GSH substrate for 10 min. The absorbance at a wavelength of 405 nm was measured by a microplate reader (Bio-Rad). A standard curve was obtained with known concentrations of GSH and used to quantify total GSH content in the unknown samples.
Statistics
The results are shown as the mean±standard deviation (SD) from at least three independent experiments. Statistical significance of the differences between two sample groups was evaluated using Student’s t-test. p<0.05 was considered statistically significant.
Go to :
Results
The protein level of MUC1-C was decreased during differentiation of mES cells
To investigate the expression patterns of MUC1 in undifferentiated mES cells and differentiated cells, we determined the mRNA and protein levels of MUC1 during RA-induced differentiation (19) of mES cells. As shown in the results of quantitative PCR, the expression level of MUC1 mRNA was significantly decreased on the first day of differentiation, recovered on day 3, and then increased on day 7 (Fig. 1A). On the other hand, the protein level of MUC1-C determined by western blotting was markedly decreased upon differentiation (Fig. 1B). FACS analysis also showed decreased expression of MUC1-C in the RA-differentiated cells compared to that of the untreated mES cells (Fig. 1C). Taken together, the results show that the expression level of MUC1-C protein in mES cells is higher than that of differentiated cells, suggesting that MUC1-C may be involved in the regulation of ES cells.
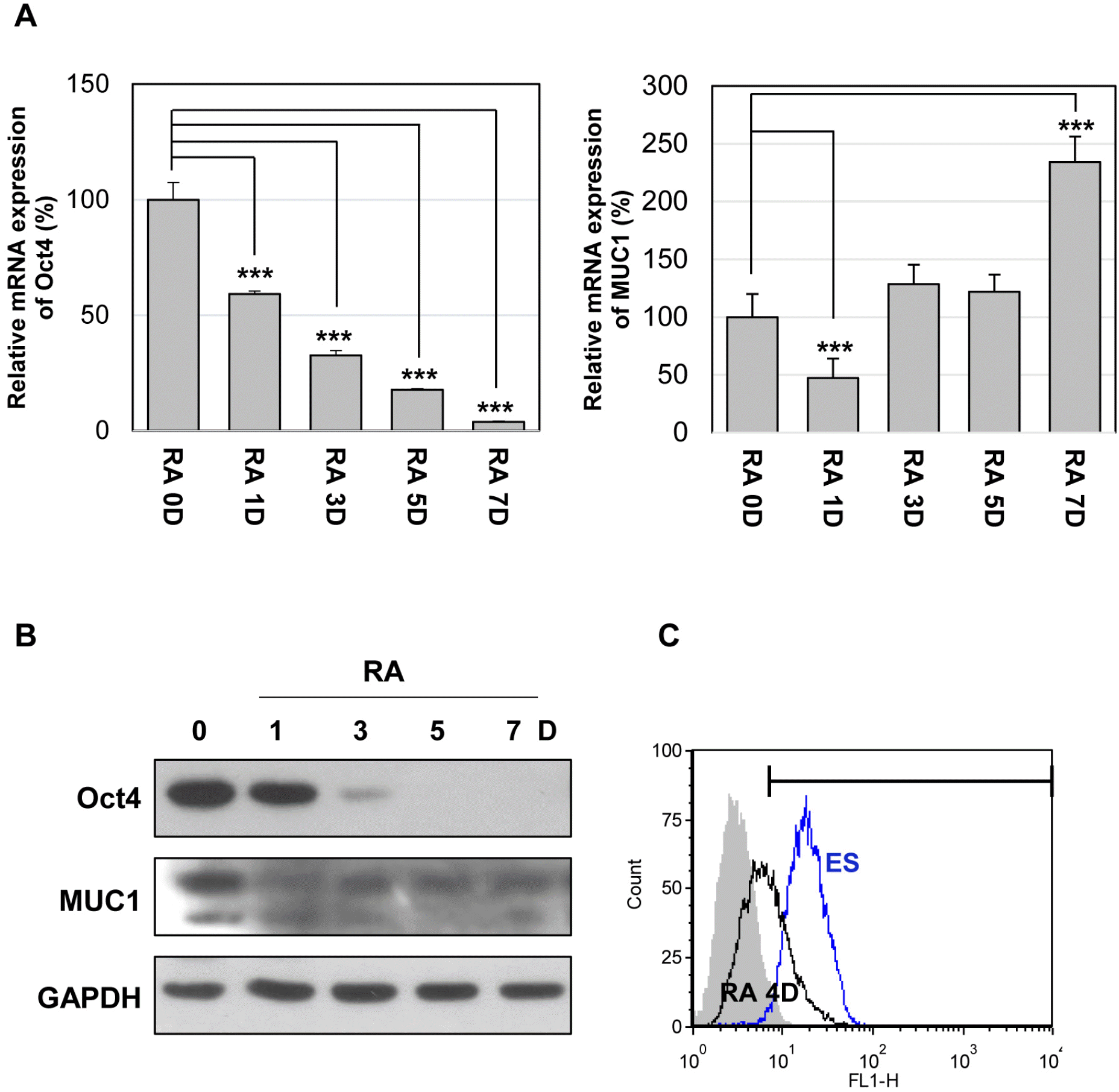 | Fig. 1Expression of MUC1 in mouse embryonic stem (mES) cells. The cells were differentiated with retinoic acid (RA, 10 μM) for the indicated period. (A) The relative expression levels of Oct4 and MUC1 mRNAs were analyzed by quantitative real-time PCR. Each bar is expressed as the mean±standard deviation (SD) of three experiments. ***p<0.001 (vs RA 0D). The protein level of MUC1-C was analyzed by western blotting (B) and flow cytometry (C).
|
Inhibition of MUC1-C reduces growth of mES cells
The MUC1 cytoplasmic tail is involved in the formation of MUC1 oligomers through a CQC motif (17). MUC1-C oligomerization directs its nuclear localization and interaction with various effectors. To investigate the potential role of MUC1-C in mES cells, we used a cell-permeable inhibitor peptide GO201, which contains the CQC motif, and a control peptide (CP1) in which CQC was substituted for AQA. To determine the effect of MUC1-C inhibition on the maintenance of mES cells, we treated the cells with GO201 or CP1 and monitored for colony formation and growth. Based on the colony size and numbers, GO201-treated cells showed reduced colony formation compared with CP1-treated cells. While CP1-treated cells retained large and well-formed ES cell colonies, GO201-treated cells displayed a concentration-de-pendent decrease in colony size and a marked increase in cell death at 20 μM (Fig. 2A).
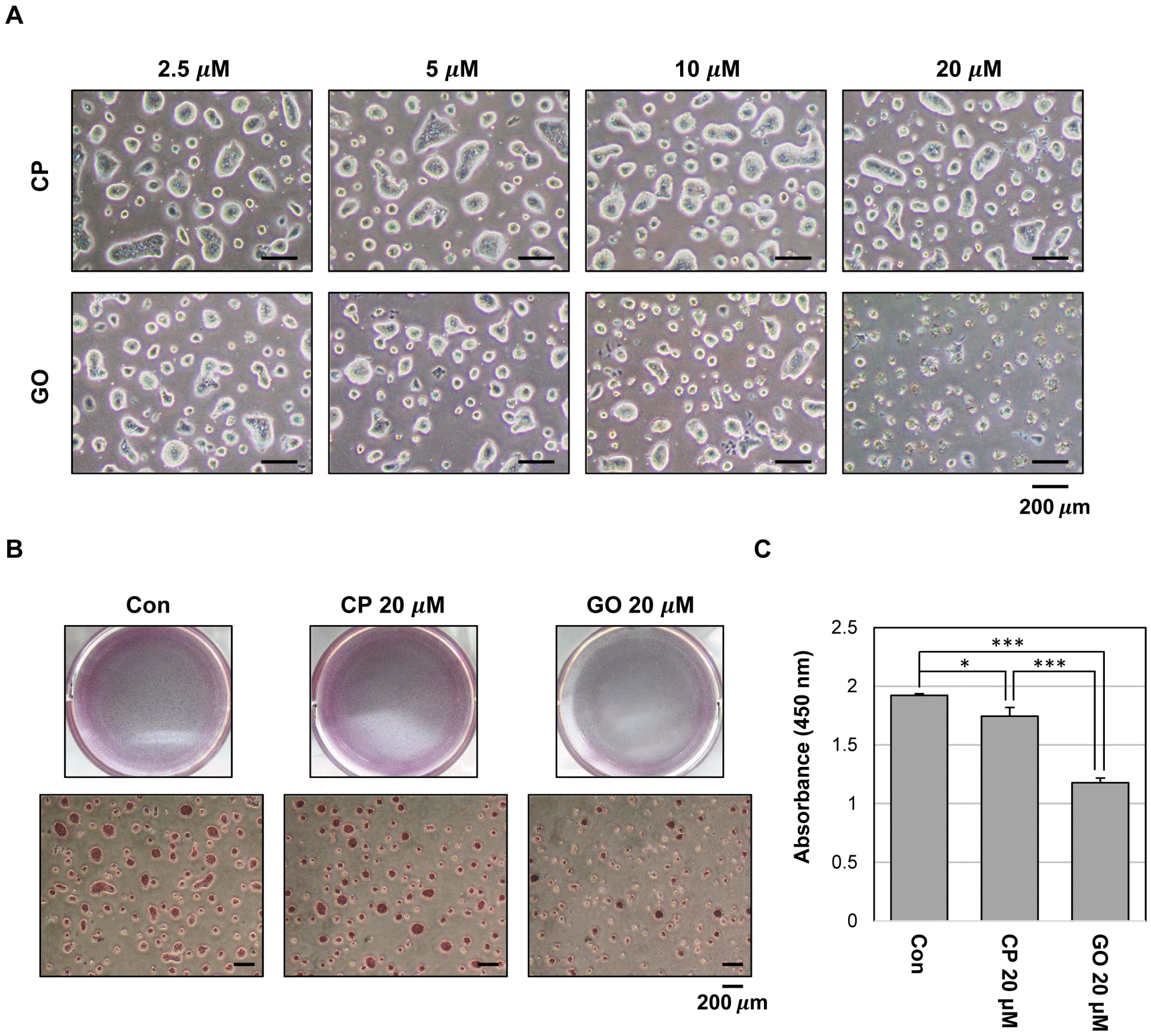 | Fig. 2Effect of MUC1-C inhibition on growth in mES cells. (A) Phase-contrast micrograph images of mES cell colonies treated with CP1 or GO201 at the indicated concentrations for 24 hr. (B) The cells were grown in the presence of CP1 or GO201 at the indicated concentration for 24 hr. Colonies were then stained with an alkaline phosphatase kit. (C) Cell proliferation assay was performed using Cell Proliferation ELISA, BrdU kit. The cells were treated with the indicated concentration of CP1 or GO201 for 24 hr. BrdU incorporation was quantified by measuring the absorbance at 450 nm using an ELISA reader. Each bar is expressed as the mean±SD of three experiments. *p<0.05, ***p< 0.001 (vs control or CP1). Con, Control; CP, CP1; GO, GO201.
|
The alkaline phosphatase assay showed that the colony size and the number of alkaline phosphatase-positive colonies were reduced by GO201 (Fig. 2B). However, the intensity of alkaline phosphatase staining showed no significant difference between the two groups. Additionally, we performed a BrdU incorporation analysis to test whether inhibition of MUC1-C affects the growth of ES cells. The results revealed that the proliferation was reduced in the 20 μM GO201-treated cells compared with the CP1-treated cells (Fig. 2C).
Inhibition of MUC1-C does not affect pluripotency of mES cells
MUC1-C activates various growth and survival pathways in a number of cancer cells (11, 13, 20). As an example, MUC1-C promotes JAK1-mediated STAT3 phosphorylation by direct interaction with JAK1 and STAT3. Consequently, MUC1-C promotes transcriptional activation of STAT3 target genes including MUC1 (11). LIF-mediated signaling pathways including the JAK/STAT pathway regulates the pluripotency of mES cells. To determine whether MUC1-C is involved in LIF signaling, we examined the effect of MUC1-C inhibitor on STAT3 activation. Serum-starved cells were incubated with CP1 or GO201 for 1 hr and then stimulated with LIF-containing medium for various periods after stimulation. As determined by western blot analysis, the degree of STAT3 phosphorylation in GO201-treated cells was similar to those of untreated cells and control peptide-treated cells up to 2 hr (Fig. 3A). Furthermore, there was no difference in the phosphorylation level of STAT3 during culture for three days in normal culture conditions with GO201 and CP1 at concentrations of 2.5 and 5 μM, which did not significantly induce cell death (Fig. 3B). Additionally, inhibition of MUC1-C did not induce changes in the mRNA and protein levels of MUC1 (Fig. 3C). These results suggest that MUC1-C is not involved in the STAT3 regulation in mES cells. Next, to examine the effect of MUC1-C inhibition on stem cell properties of mES cells, we examined expression levels of Oct4 as a pluripotency marker. There was no change in the expression level of Oct4 after treatment with GO201 or CP1 (Fig. 3D). These results suggest that the inhibition of MUC1-C in mES cells reduces cell growth without affecting the pluripotency marker expression and LIF signaling, at least within the examined time periods.
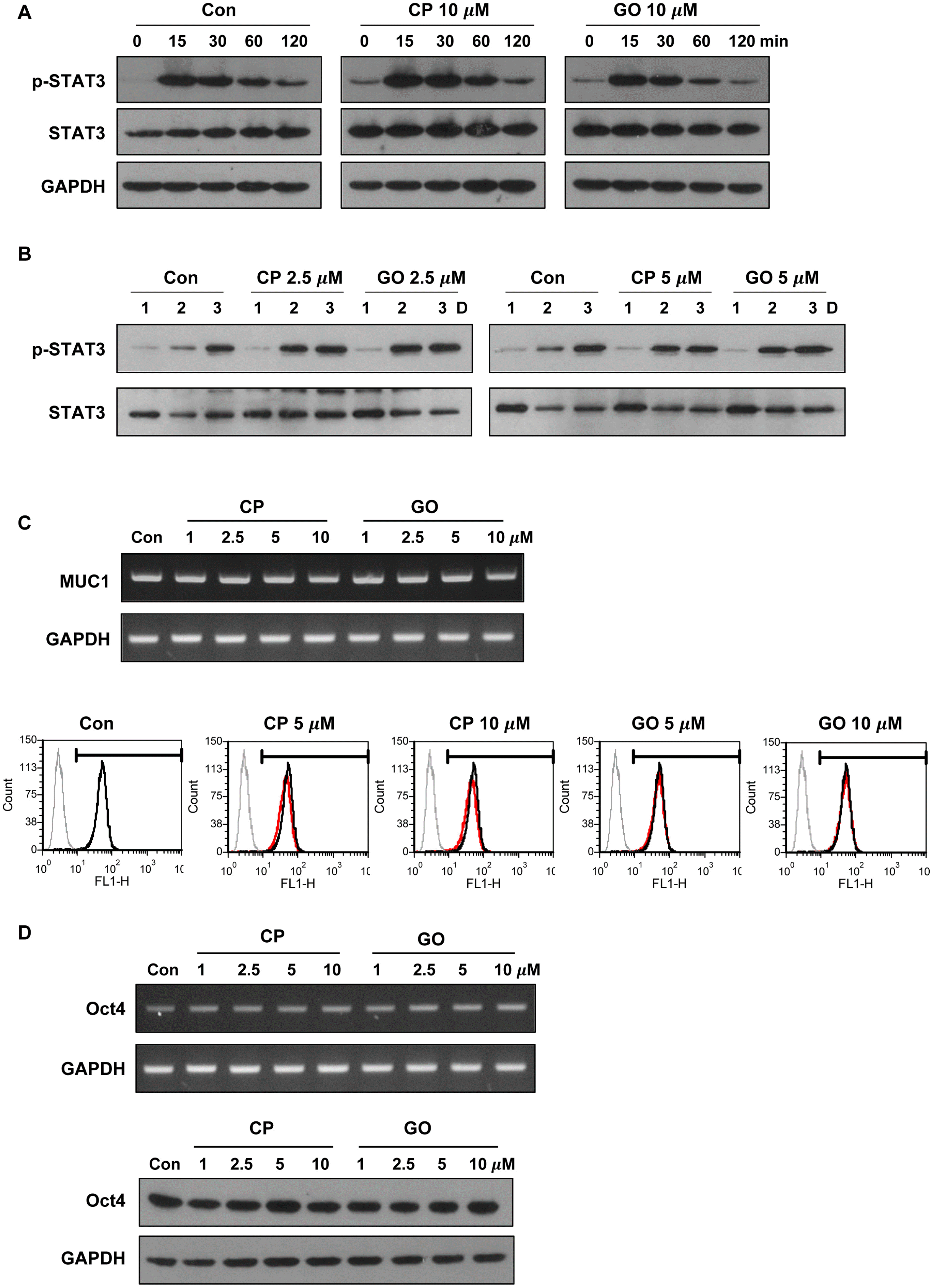 | Fig. 3Effect of MUC1-C inhibition on STAT3 signaling and expression of pluripotency marker in mES cells. (A) The cells were serum starved overnight and then preincubated for 1 hr in the presence of CP1 or GO201 (10 μM). They were then stimulated with FBS (15%) and LIF for the indicated period and analyzed by western blotting. (B) The cells were treated with CP1 or GO201 at the indicated concentrations for three days and analyzed by western blotting. (C) The cells were treated with CP1 or GO201 at the indicated concentrations for 24 hr. Expression levels of MUC1 mRNA and protein were analyzed by RT-PCR (upper) and flow cytometry (lower). The histogram peak represents the expression of MUC1-C: Grey line, negative control; Black line, untreated control; Red line, CP1- or GO201-treated samples. (D) The cells were treated with CP1 or GO201 at the indicated concentrations for 24 hr. Expression levels of Oct4 mRNA and protein were analyzed by RT-PCR (upper) and western blotting (lower). Con, Control; CP, CP1; GO, GO201.
|
Inhibition of MUC1-C induces cell cycle arrest and promotes cell death in mES cells
ES cells have a specific cell cycle profile, with a short G1 phase and a high proportion of cells in S phase, which allows rapid division and proliferation of ES cells (21). To identify the mechanism of the cell growth inhibition, we performed a cell cycle analysis using the CP1- or GO201-treated mES cells. The treatment with 2.5 μM GO201 increased the cell population in the G1 phase and reduced the cell population in the S phase (Fig. 4A). To determine whether decreased cell growth is associated with increased apoptosis of mES cells, we performed annexin V/PI staining and found that GO201 treatment increased cell death and apoptosis of mES cells in a concentration-dependent manner (Fig. 4B). The treatment with 20 μM GO201 induced a 10-fold and three-fold increase of dead cell and apoptotic cell populations, respectively, compared with 20 μM CP1 treatment. In addition, GO201 reduced the expression of Mcl1 and Bcl2, known as anti-apoptotic factors (Fig. 4C). These results show that the inhibition of MUC1-C induces cell cycle change and increases cell death along with reduced anti-apoptotic factor expression in mES cells, suggesting that MUC1-C may contribute to the sustained growth and survival of mES cells.
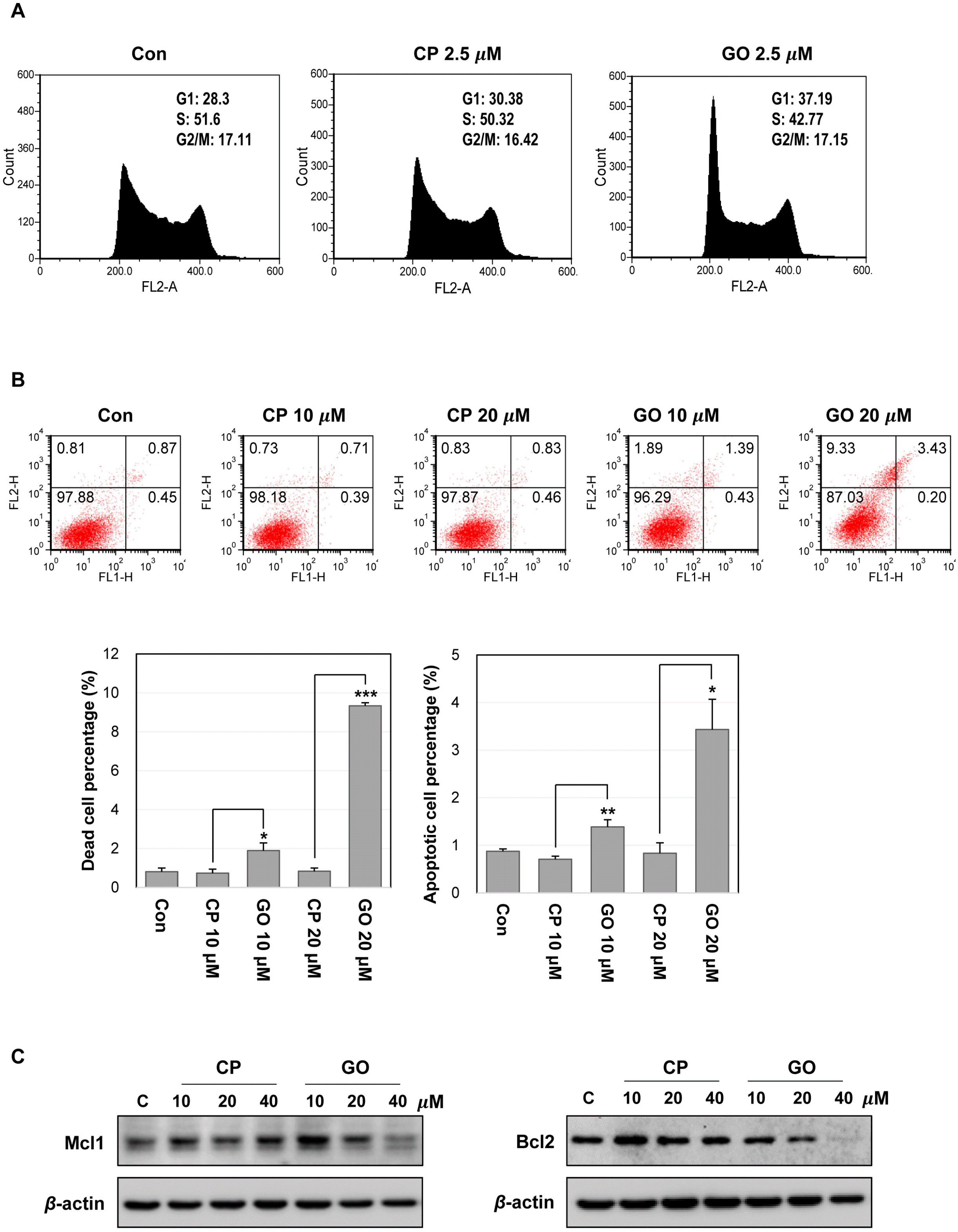 | Fig. 4Inhibition of MUC1-C induces cell cycle changes and cell death in mES cells. (A) Cell cycle analysis of mES cells by flow cyto-metry. The cells were treated with the indicated concentration of CP1 or GO201 for 24 hr. (B) Apoptosis of mES cells treated with CP1 or GO201 for 24 hr was assessed by Annexin V/PI staining. Each bar is expressed as the mean±SD of three experiments. *p<0.05, **p<0.01, ***p<0.001 (vs CP1). The results at the bottom are shown as percentages of dead cell and apoptotic cell population. (C) Expression levels of Mcl1 and Bcl2 were analyzed by western blotting, after treatment with CP1 or GO201 at the indicated concentrations for 24 hr. Con, Control; CP, CP1; GO, GO201.
|
Inhibition of MUC1-C disrupts ROS balance in mES cells
Previous studies have shown that the overexpression of MUC1 decreases intracellular ROS levels in various cancer cell lines (22). The balance of redox is one of the important factors in maintaining the characteristics of ES cells. Therefore, the change in the basal ROS level may be an important indicator for the functional state of ES cells. In this regard, ROS levels of GO201- or CP1-treated mES cells were assessed and the results showed that the treatment with GO201 but not CP1 increased the cellular ROS level in a concentration-dependent manner (Fig. 5A). Generally, an increased ROS level is associated with a decrease in intracellular GSH level. There was a slight increase of the GSH level in the CP-1-treated cells and the reason for this is not clear. However, the GSH level was significantly decreased by the treatment with GO201 (Fig. 5B). To further support MUC1-mediated ROS control, we tested whether the increased ROS level by GO201 treatment is restored by adding NAC, an antioxidant. As shown in Fig. 5C, the ROS level was not increased by GO201 in the presence of NAC. Furthermore, 20 μM GO201-treated mES cells showed a concentration-dependent decrease of cell death and increase of colony size when NAC was added (Fig. 5D). Therefore, these results suggest that the growth inhibition and cell cycle arrest induced by inhibition of MUC1-C is associated with the change of the redox balance in mES cells.
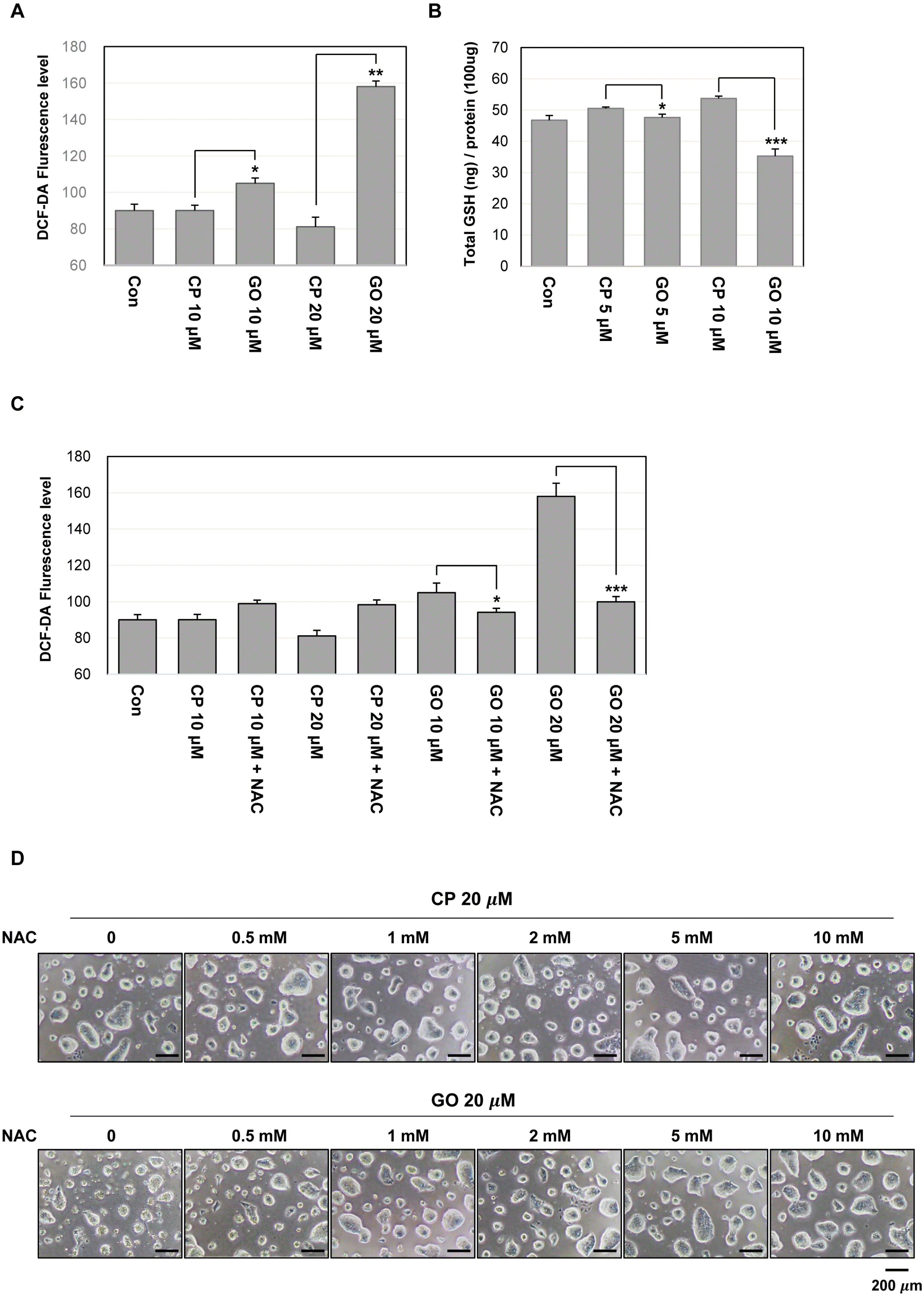 | Fig. 5MUC1-C inhibition increases ROS level in mES cells. (A) ROS level measured by flow cytometry using DCF-DA. The cells were treated with the indicated concentrations of GO201 or CP1 for 24 hr, incubated with 10 μM of DCF-DA for 30 min, and analyzed by flow cytometry. (B) The cells were treated with the indicated concentrations of GO201 or CP1 for 24 hr. The cell lysates were incubated with glutathione reductase and the GSH substrate, and the absorbance values were measured at a wavelength of 405 nm. (C) The cells were untreated or treated with 10 μM of NAC and were treated with the indicated concentrations of GO201 or CP1 for 24 hr. The cells were then incubated with 10 μM of DCF-DA for 30 min and analyzed by flow cytometry. Each bar is expressed as the mean±SD of three experiments. *p<0.05, **p<0.01, ***p<0.001 (vs CP1). (D) Images of mES cell colonies. The cells were untreated or treated with the indicated concentrations of NAC and were treated with 20 μM of GO201 or CP1 for 24 hr. Con, Control; CP, CP1; GO, GO201.
|
Go to :
Discussion
MUC1 is a membrane-associated glycosylated protein, which is expressed in several types of normal and cancerous cells. In particular, MUC1-C is overexpressed in various cancer cells, which regulates adhesion, migration, and intracellular signaling (23-28). Considering that pluripotent cells have common properties with cancer cells, it is intriguing to investigate the relevance of MUC1-C in ES cells. In this study, we investigated possible implication of MUC1-C in mES cells. We identified that MUC1-C is expressed in self-renewing mES cells and is decreased during differentiation induced by retinoic acid. These results suggest that MUC1-C may play a specific role in the maintenance of undifferentiated ES cells. This is consistent with the finding that the expression level of cleavage product MUC1* is higher than that of full-length MUC1 in undifferentiated human ES cells, and it decreases as ES cell differentiation progresses (29). The human and mouse MUC1 protein sequences have only about 30% identity; the C-terminal cytoplasmic domains of MUC1, however, are highly homologous with more than 85% identity (30). Therefore, we expected that MUC1-C plays some role in the growth and maintenance of ES cells.
The results of previous studies on MUC1-C in cancer cells show that it mediates multiple roles through interaction with several signaling factors such as STAT3 (11), p53 (31), and β-catenin (32). To investigate the functional role of MUC1-C in mES cells, we used a cell-penetrating inhibitor peptide GO201. We found that inhibition of MUC1-C with GO201 significantly reduced the growth of mES cells and decreased the number of alkaline phosphatase-positive colonies. The BrdU incorporation analysis revealed that the proliferation of mES cells was significantly reduced by MUC1-C inhibition. To determine whether inhibition of MUC1-C affects pluripotency, we analyzed changes in specific markers of mES cells. LIF activates multiple signaling pathways such as JAK/STAT3, PI3K/AKT, and MAPK in mES cells. Oct4 is a homeodomain transcription factor of the POU family and is essential for the self-renewal of undifferentiated embryonic stem cells (33). Inhibition of MUC1-C did not induce a change in STAT3 signaling, a representative signaling pathway of mES cells, and did not affect the expression of Oct4. These results suggest that MUC1-C may be involved in cell proliferation rather than the maintenance of pluripotency in mES cells.
Our next question was how inhibition of MUC1-C reduces mES cell growth. Previous studies have demonstrated that MUC1-C protects cells from cell death induced by various stresses (22, 34, 35). Furthermore, it is known that MUC1-C contributes to maintaining intracellular redox balance in cancer cells. For example, Yin et al. (36) reported that MUC1-C increases the expression of antioxidant enzymes in several cancer cell lines, which protects cells from oxidative stress-induced cell death. Raina et al. (17) reported that inhibition of MUC1-C interferes with redox balance and activates a DNA damage response in human breast cancer cells. Furthermore, a number of recent studies have shown that MUC1-C regulates the expression of transcription factors involved in metabolic processes in cancer cells. Kosugi et al. (37) reported that MUC1-C binds directly to pyruvate kinase M2 (PKM2), a key enzyme in cancer metabolism, and regulates PKM2 activity. Chaika et al. (38) reported that MUC1-C induces glucose uptake and glycolysis through HIF-1α regulation, and directly occupies multiple promoters of the glycolytic genes in pancreatic cancer cells. MUC1-C expression also increases the expression of glycolytic genes including hexokinase 2 (HK2) (38). Consistent with the results from cancer cells, inhibition of MUC1-C in mES cells induced an increase in intracellular ROS level along with a decrease in GSH level and increased cell death. Therefore, it is likely that MUC1-C plays a role in maintaining ROS levels in ES cells under normal conditions. However, inhibition of MUC1-C in mES cells did not affect the stability of HIF-1α (data not shown). Although HK2 and PKM2 genes are known to regulate metabolic processes in ES cells (39), the mRNA expression levels of these genes were not changed by MUC1-C inhibition in mES cells (data not shown). These results suggest that MUC1-C has an ES cell-specific mechanism regulating metabolic processes. Further study on the functional role of MUC1-C will give more information regarding ES cell self-renewal and optimal conditions for the stable and efficient maintenance of ES cells.
Go to :
 XML Download
XML Download