

 PDF
PDF Citation
Citation Print
Print


Introduction
Many studies have emphasized the clinical importance of mesenchymal stem cells (MSCs) in degenerative and regenerative medicine. Recently, extensive clinical trials of MSCs have been conducted. However, their efficacy depreciates because of poor survival and engraftment in injured organs. In particular, the therapeutic mechanism or “mode of action” is lacking. MSC-based therapy using a gene delivery system shows enhanced efficacy due to improvement in cell survival and secretion of numerous growth factors (1). In general, viral transfection has advantages such as high efficiency in genetic transfer and stable gene expression. Previously, an optimal protocol was developed using lentiviral transfection in bone marrow-derived MSCs (BM-MSCs), resulting in higher efficiency and lower cell toxicity (2, 3). However, viral vectors have several limitations, including their risk of immunogenicity and chromosomal integration in clinical applications (4). The current strategy, utilizing NucleofectionTM for the nonviral gene delivery, is not only acceptable with regard to safety issues but also is easy to handle, indicating possible applications in patient. In particular, this method has been successfully applied to difficult-to-transfect primary cells, including MSCs. NucleofectionTM platforms could potentially affect the innate abilities of MSCs (5). In our previous reports, naïve placenta-derived MSCs (PD-MSCs) were transfected using NucleofectionTM and subjected to genetic analyses (6). However, the NucleofectionTM nonviral gene delivery in MSCs is still being developed, requiring their optimization to safely establish a cell line, maintain the characteristics of naïve MSCs, and promote therapeutic efficacy. In this report, we will highlight the optimal protocol for the generation of functionally enhanced MSCs using a nonviral gene delivery system.
Materials and Methods
Primary cells
Naïve PD-MSCs were isolated as previously described using placentas for all participating women at term (≥37 gestational weeks) (7). The protocol was approved by the Institutional Review Board (IRB) of CHA General Hospital, Seoul, Republic of Korea (IRB 07-18).
►Note: Naïve PD-MSCs were maintained in alpha-modified minimum essential medium (α-MEM; HyClone, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA), 1% penicillin/streptomycin (P/S; Gibco), 25 ng/ml human fibroblast growth factor-4 (hFGF- 4; PeproTech, USA), and 1 μg/ml heparin (Sigma- Aldrich, USA).
Reagents
▪ α-MEM (cat. no. SH30265.01)
▪ FBS (cat. no. GIB-16000-044)
▪ hFGF4 (cat. no. 100-31-100)
▪ Heparin (cat. no. H3149)
▪ P/S (cat. no. GIB-15140-122)
▪ Dulbecco’s phosphate buffered saline (DPBS; Wellgene, cat. no. LB001-02, Korea)
▪ Phosphatase of regenerating liver-1 (PRL-1) plasmid (Origene, Inc., cat. no. RG200435, USA)
▪ pCMV-AC-GFP (Origene, Inc., cat. no. PS100010)
▪ TrypLETM Express (+) Phenol Red (Gibco, cat. no. GIB-12605-010)
▪ Trypan blue, 0.4% (Gibco, cat. no. GIB-15250-061)
▪ P1 Primary Cell 4D NucleofectorTM X Kit L (Lonza, cat. no. V4XP-1024, Switzerland)
►Note: Including P1 Primary Cell NucleofectorTM Solution, Supplement 1, 50 μg
pmaxGFPTM Vector (1 μg/μl in 10 mM Tris pH 8.0)
▪ Neomycin (Sigma-Aldrich, cat. no. N6386)
Equipment
▪ 4D-NucleofectorTM Core Unit (Lonza, cat. no. AAF- 1002B)
▪ 4D-NucleofectorTM X Unit (Lonza, cat. no. AAF-1002X)
▪ NucleocuvetteTM Vessels (Lonza, cat. no. AXP-1003)
▪ T75 cell culture flask (SPL, cat. no. 70075, Korea)
▪ 100 mm cell culture dish (Nunc, cat. no. 150466, Denmark)
▪ Hemocytometer (Marienfeld, cat. no. 0610039094, Germany)
▪ Conical tube, 15 ml (SPL, cat. no. 50015, Korea)
▪ Centrifuge (Labogene, cat. no. 1730R, Korea)
▪ CO2 incubator (Thermo Scientific, Hera Cell, cat. no. H150-CO2, USA)
Pre NucleofectionTM: 1.1 Seed naïve PD-MSCs (2.5× 105 cells) in a T75 cell culture flask containing 15 ml of cell culture medium and incubate overnight at 37℃ in a 5% CO2 incubator.
1.2 Naïve PD-MSCs (passage<9) should reach approximately 80% confluence.
►Note: Transfection using naïve PD-MSCs may be donor-dependent. Thus, it is recommended to use passages earlier than 9.
1.3 Remove the medium from the cultured naïve PD- MSCs and wash the cells with DPBS 2 times.
1.4 Add 2 ml of TrypLETM Express (+) Phenol Red and incubate the cells for 2 min at 37℃ in a 5% CO2 incubator.
1.5 Harvest cells with prewarmed complete medium and obtain an aliquot of cells.
1.6 Count the Trypan blue-stained cells using hemocytometer and divide the cell suspension (5×105 cells/tube) in each 15 ml conical tube.
1.7 Centrifuge at 200 x g for 10 min at room temperature.
►Note: To avoid the cell damage, speed should not exceed 200 x g.
NucleofectionTM: 2.1 Ensure that the entire supplement solution is added to the NucleofectionTM solution.
►Note: The ratio of the NucleofectionTM solution to the supplement solution is 4.5:1. The mixture is stable for 3 months at 4℃. For a single reaction, mix 82 μl of NucleofectionTM solution and 18 μl of supplement solution.
2.2 Operate the 4D NucleofectorTM System and set the appropriate NucleofectorTM program.
►Note: Turn on the 4D NucleofectorTM System → choose a X unit device → choose a cuvette vessel → choose the number of cuvettes → Select a cell type program “MSC, human” → pulse code was set up as FF-104 → press OK
►Note: Pulse code as FF-104 for high efficiency.
2.3 Prepare complete medium in 100 mm cell culture dish and preincubate/equilibrate each plate without cells at 37℃ in a 5% CO2 incubator.
►Note: For the stabilization of transfected PD-MSCs, its step is important to improve transfection efficiency and viability.
2.4 After centrifugation at 200 x g for 10 min at room temperature, carefully resuspend the cell pellet in 100 μl of NucleofectionTM solution.
2.5 Add an appropriate volume of plasmid in each tube and mix well.
►Note: pmaxGFPTM Vector (2 μg/5×105 cells/100 μl NucleofectionTM solution 100 μl/cuvette).
►Note: PRL-1/pCMV-AC-GFP (5 μg/5×105 cells/100 μl NucleofectionTM solution 100 μl/cuvette).
►Note: Make sure that matermixtures must mix well while pipetting.
►Note: Volume of mastermixtures should not exceed maximum 10 % of the total reaction volume (10 μl for 100 μl reaction). It is recommended that the concentration of plasmid is suitable for 1 μg/μl.
2.6 Transfer mastermixtures into the NucleocuvetteTM Vessels and place NucleocuvetteTM Vessels in the retainer of the 4D NucleofectorTM X unit device.
►Note: In order to prevent an electrical signal error, avoid air bubbles while transferring mastermixtures.
2.7 Start Nucleofection process according to the presetting value (refer to 2.2).
2.8 After running, remove NucleocuvetteTM Vessels in the retainer of the device and incubate NucleocuvetteTM for 10 min at room temperature.
2.9 Add 500 μl of prewarmed PD-MSCs medium and gently resuspend the transfected cells.
2.10 Plate the transfected cells in preincubated cell culture plates.
Post NucleofectionTM: 3.1 Incubate the transfected cells at 37℃ in a 5% CO2 incubator for 24 h.
►Note: Gene expression is often detectable after 4∼8 h. However, gene or GFP expression in PD-MSCs recommended 24 h post NucleofectionTM.
3.2 After 24 h, remove medum and change selection medium including 1.5 mg/ml neomycin for 7 days.
3.3 Confirm the gene or GFP expression.
Characterizations
Cell viability and transfection efficiency using quantitative real-time polymerase chain reaction (qRT-PCR) and western blotting: PRL-1-overexpressing PD-MSCs (PD-MSCsPRL-1, PRL-1+) were counted 24 h post transfection to examine the transfected cell viability. For analysis of the efficiency of PD-MSCsPRL-1 post-transfection, total RNA and protein lysate were extracted from the cell pellet. qRT-PCR analysis was performed with primers (Table 1) using the CFX ConnectTM Real-Time System (Bio-Rad, USA). Western blotting was performed with primary monoclonal anti-turboGFP (1:500, Origene, Inc.) and horseradish peroxidase (HRP)-conjugated anti-goat IgG (Santa Cruz Biotechnology, USA).
Expression of stemness markers by reverse transcription (RT)-PCR: For analysis of stemness markers in PD-MSCsPRL-1, PCR amplification was conducted with Taq DNA polymerase (Solgent, Korea) according to the manufacturer’s instructions. Each primer is provided in Table 1.
Immunophenotypes by flow cytometry: For analysis of the immunophenotyping of cell surface antigens, PD- MSCsPRL-1 cells were stained with phycoerythrin (PE)- conjugated CD13, CD90, and CD105 (BD Bioscience, USA) and fluorescein isothiocyanate (FITC)-conjugated HLA-ABC, HLA-DR (BD Bioscience), and HLA-G (Abcam, UK) using a CytoFLEX flow cytometer (Beckman Coulter, USA). All data sets were collected from 10,000 cells.
Differentiation potential: Naïve PD-MSCs were previously confirmed for their multilineage differentiation ability (8). PD-MSCsPRL-1 were plated (5×103 cells/cm2) in differentiation induction medium using a StemPro Adipogenesis and Osteogenesis differentiation kit (Gibco) as well as hepatogenic differentiation as previously described (9). After each differentiation, cells were stained with oil red O (Sigma-Aldrich) for adipogenesis, von Kossa (Sigma-Aldrich) for osteogenesis, and 1 mg/ml indocyanine green (ICG) (Dong in Dang Pharm., Korea) for uptake.
Karyotyping: For analysis of karyotypes of naïve and PD-MSCsPRL-1, G-banding techniques were performed, and the karyotype was elucidated by the International System for Human Cytogenomic Nomenclature (ISCN 2016) using an Axipskop 2 plus light microscope (Zeiss, Germany).
Teratoma formation: For analysis of teratoma formation by PD-MSCsPRL-1, 5×105 cells were transplanted into one testis (Tx; n=2) while another testis was not injected (Con; n=2) in nine-week-old male nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice (Laboratory Animal Research Center, Bundang CHA Medical Center, Seongnam, Korea). After 14 weeks, the mice were sacrificed, testis tissues were stained with hematoxylin & eosin (H&E), and qRT-PCR analysis was performed with the human Alu sequence (Table 1).
Results
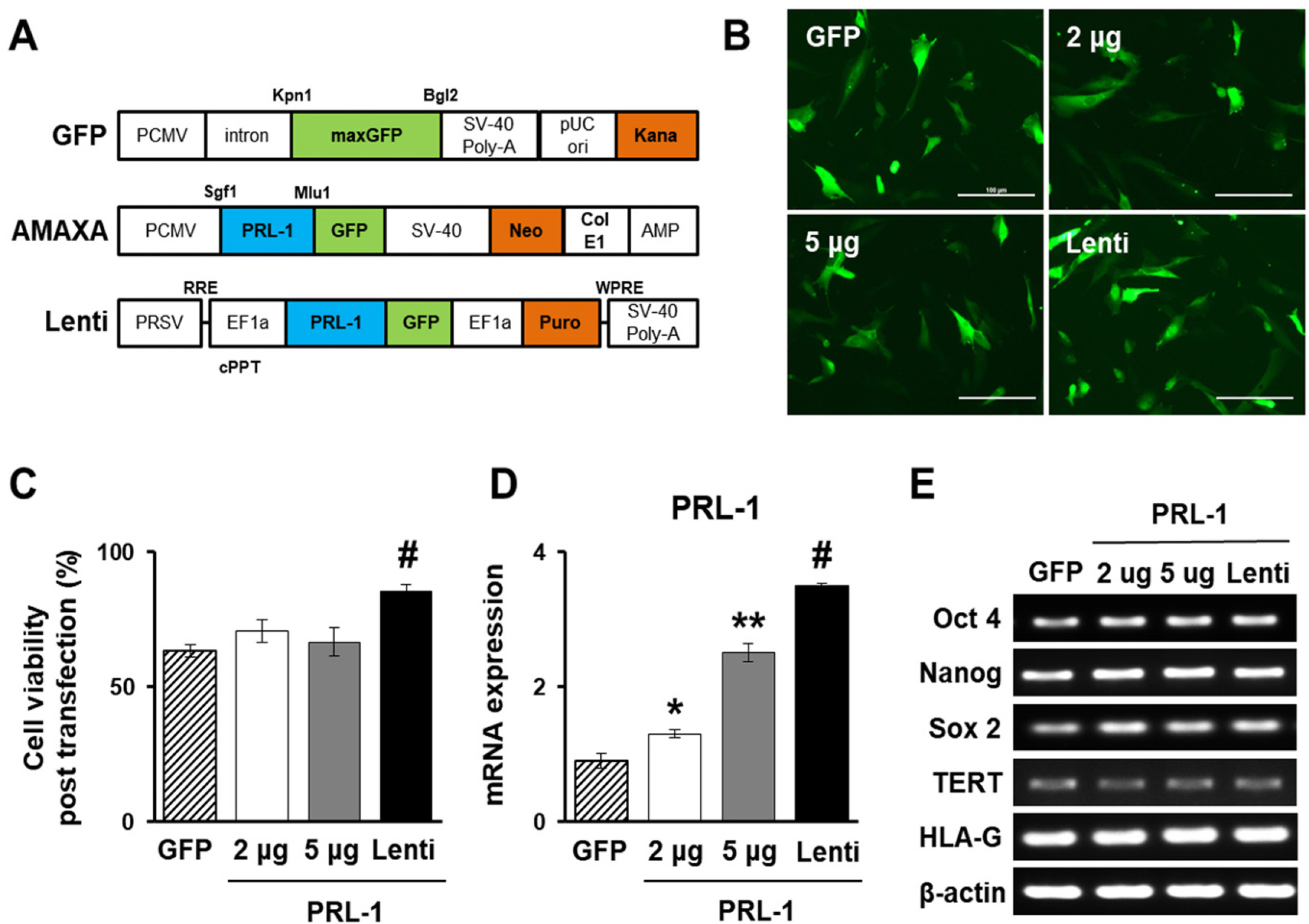
Optimization of transfection in PD-MSCs
Naïve PD-MSCs were transfected using the nonviral AMAXA electroporation technique as well as the lentiviral system as a positive control group for PRL-1 gene expression (Fig. 1A). To optimize the concentration of PRL-1 plasmid using nonviral electroporation, we used both 2 and 5 μg, and GFP positivity was indicated (Fig. 1B). Cell viability in the 2 and 5 μg groups was not significantly different (Fig. 1C). mRNA expression of PRL- 1-overexpressing PD-MSCs (PD-MSCsPRL-1, PRL-1+) using both gene delivery systems was increased. In electroporation system, 5 μg of PRL-1 resulted in significantly higher effects than 2 μg of PRL-1 (Fig. 1D) as well as PRL-1-overexpressing bone marrow-derived MSCs (BM- MSCsPRL-1) (Supplementary Fig. S1). Additionally, stemness markers (e.g., Oct4, Nanog, Sox2), telomerase reverse transcriptase (TERT), and immunomodulatory factor HLA-G expression was maintained regardless of the concentration of the PRL-1 plasmid (Fig. 1E). Therefore, these data suggest that 5 μg of transfected PD-MSCsPRL-1 using a nonviral system is as efficient as the lentiviral system.
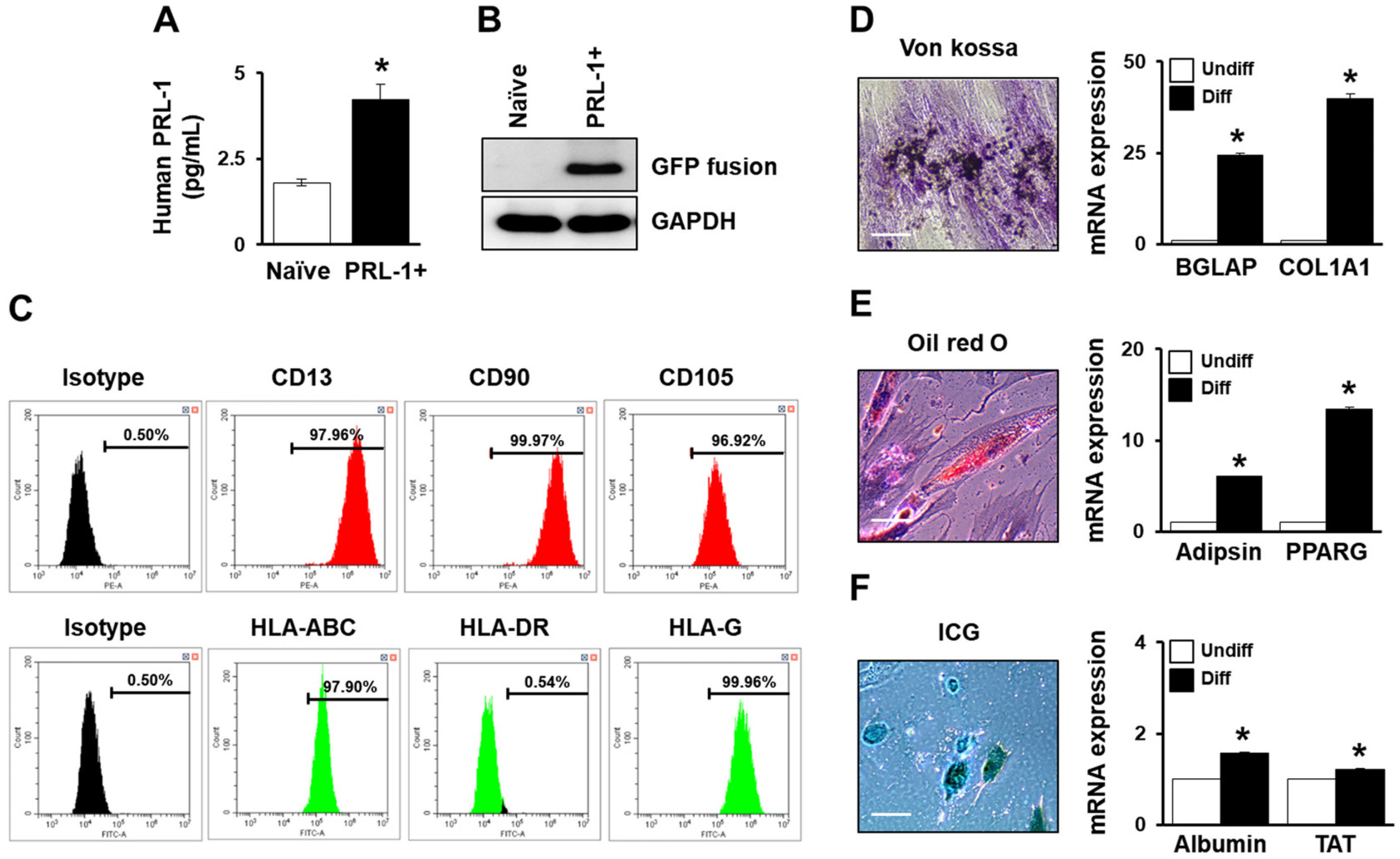
Characterization of PD-MSCsPRL-1 using a nonviral electroporation system
In the cultured supernatant, the secreted PRL-1 level was significantly higher than that in naïve cells (Fig. 2A). Additionally, PRL-1 transfection with the GFP fusion protein was confirmed (Fig. 2B). PD-MSCsPRL-1 were confirmed by surface markers in flow cytometry. Similar to the naïve patterns, the expression of MSC markers (e.g., CD13, CD90, CD105, HLA-ABC, and HLA-G) in PD-MSCsPRL-1 was positive, whereas HLA-DR was negative (Fig. 2C). To analyze the differentiation abilities of PD-MSCsPRL-1, we confirmed mesodermal (osteogenic and adipogenic) and endodermal (hepatogenic) lineages using von Kossa, oil red O, and ICG staining, respectively (Fig. 2D∼F). In addition, the expression of osteogenic (e.g., bone gamma carboxyglutamic acid containing protein; BGLAP and collagen type 1 alpha 1; COL1A1), adipogenic (e.g., adipsin and peroxisome proliferator activated receptor gamma; PPARG), and hepatogenic (e.g., albumin and tyrosine aminotransferase; TAT) markers in differentiated PD-MSCsPRL-1 were significantly increased compared to those of the undifferentiated PD-MSCsPRL-1 as shown by qRT-PCR analysis (*p<0.05). Thus, these findings indicate that PD-MSCsPRL-1 retain the potential to differentiate into multiple lineages.
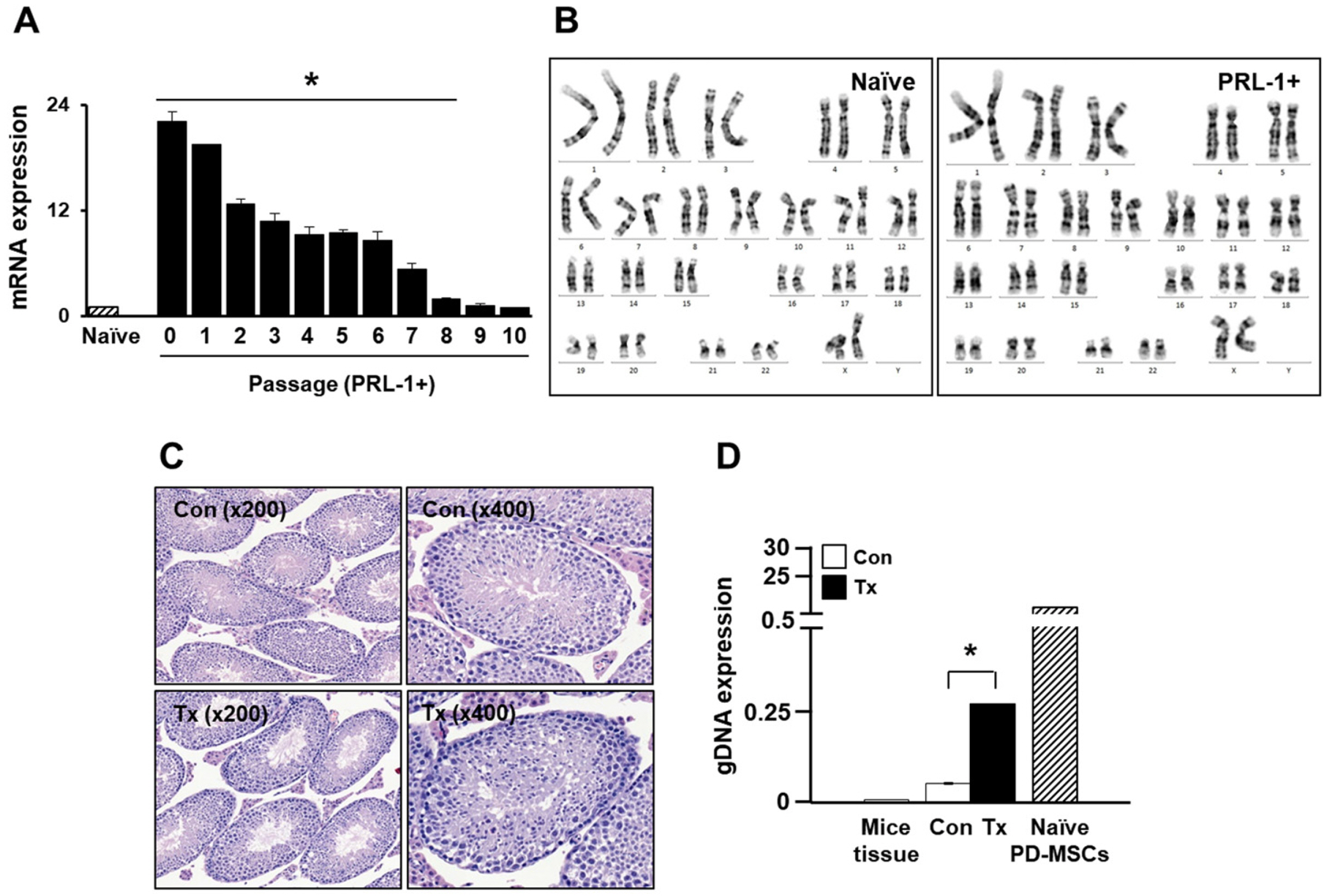
Safety evaluation of PD-MSCsPRL-1 using the nonviral electroporation technique
The expression of PRL-1 in PD-MSCsPRL-1 was significantly increased until passage 8 compared to that of the naïve PD-MSCs (Fig. 3A, *p<0.05). The karyotypes of the naïve and PD-MSCsPRL-1 were diploid and stable (Fig. 3B). In NOD/SCID mouse testes, there was no teratoma formation (Fig. 3C), and human Alu expression was significantly increased after PD-MSCPRL-1 transplantation (Fig. 3D, *p<0.05). Therefore, these data suggest that PD-MSCsPRL-1 generated by a nonviral electroporation system showed efficacy as well as safety.
Conclusion
An optimized protocol for PD-MSCsPRL-1 using nonviral AMAXA electroporation was established, and its efficiency was confirmed. In addition, the characteristics of PD-MSCsPRL-1 were similar to those of the naïve MSCs, and a safety evaluation was accomplished. Therefore, this nonviral electroporation technique could be a new strategy for the clinical application of stem cell therapy.
Supplementary Materials
Supplementary data including one figure can be found with this article online at https://doi.org/10.15283/ijsc20117
 XML Download
XML Download