

 PDF
PDF Citation
Citation Print
Print


Introduction
Fanconi Anemia (FA) is a genetic disorder associated with bone marrow failure and high risk of cancer particularly leukemia (1–3). The disease is caused by defects in any of at least 22 genes (FANCA-W), which together constitute the so-called FA pathway engaged in some of the most important cellular processes such as DNA replication, cell-cycle control, and DNA damage response and repair (4–6). In the context of hematologic malignancies, FA commonly progresses from bone marrow (BM) failure to a pre-leukemic myelodysplastic syndrome (MDS) stage and finally evolves to acute myeloid leukemia (AML). Patients with FA commonly develop pancytopenia during the first few years of life, and have high susceptibility of developing MDS or AML (7, 8). Currently, hematopoietic stem cell (HSC) transplantation is considered the best treatment for BM failure and leukemia in FA patients (9, 10).
It is postulated that the FA pathway promotes the error-free homologous recombination (HR) repair pathway while suppressing the error-prone non-homologous end-joining (NHEJ) pathway (11–14). In supporting this notion, recent studies have shown that FA deficiency enhanced the error-prone NHEJ repair, leading to increased genomic instability, and that genetic or pharmacological inhibition of the NHEJ pathway could rescue the FA phenotype (11, 13). However, the biological consequence on the interplay between FA pathway and the NHEJ pathway is not clear. In this study, we show that the NHEJ activity of DNA-PKcs is required for DNA damage-induced expansion of Fanca−/− pre-leukemic HSCs.
Go to : 
Materials and Methods
Mice and treatment
Fanca−/− mice were generated by interbreeding the heterozygous Fanca+/− (C57BL/6:B6, CD45.2; Dr. Madeleine Carreau at Laval University). DNA-PKcs3A/3A mice (C57BL/6: B6, CD45.2; provided by Dr. Benjamin P. C. Chen at University of Texas Southwestern Medical Center) (15) were generated by interbreeding heterozygous DNA-PKcs+/3A mice. These mice were C57BL/6:B6, CD45.1 or 2 All the animals including BoyJ mice (C57BL/6:B6, CD45.1) were maintained in the animal barrier facility at Cincinnati Children’s Hospital Medical Center. For in vivo mitomycin C (MMC; Sigma-Aldrich, St. Louis, MO) treatment, mice were intraperitoneal (i.p.) injected with 0.3 mg/kg of MMC weekly for consecutive 6 weeks (16). All animal experiments were performed in accordance with the institutional guidelines and approved by the Institutional Animal Care and Use Committee of Cincinnati Children’s Hospital Medical Center (IACUC2018-0006).
Isolation of bone marrow cells and flow cytometry analysis
The femora and tibiae were harvested from the mice immediately after their sacrifice with CO2. Bone marrow (BM) cells were flushed from bones into Iscove’s modified Dulbecco’s medium (IMDM; Invitrogen) containing 10% FCS, using a 21-guage needle and syringe. Low-density BM mononuclear cells (LDBMMNCs) were separated by Ficoll Hypaque density gradient (Sigma-Aldrich, St. Louis, MO) and washed with IMDM medium.
For flow analysis and cell sorting, the lineage marker (Lin) mixture (BD Biosciences, San Jose, CA) for BM cells from treated or untreated mice included the following biotinylated antibodies: CD3ɛ (145-2C11), CD11b (M1/70), CD45R/B220 (RA3-6B2), mouse erythroid cells Ly-76 (Ter119), Ly6G and Ly-6C (RB6-8C5). Other conjugated antibodies (BD sciences, San Jose, CA) used for surface staining included: CD45.1 (A20), CD45.2 (A104), Sca1 (D7), c-kit (2B8), CD48 (HM48-1), CD150 (9D1). For the cell sorting, lineage negative cells were enriched using lineage depletion reagents (StemCell Technologies) according to the manufacturer’s instruction. The Lin-negative and LSK populations were acquired by using the FACS-Aria II sorter (BD Biosciences).
Bone marrow transplantation (BMT)
For determination of MMC-induced expansion of the Fanca−/− HSCs, 2,000 LSK cells, along with 200,000 c-Kit-depleted protector cells, were transplanted into lethally irradiated BoyJ (CD45.1+) recipients. The recipients were then i.p. injected with low dose of MMC (0.3 mg/kg) weekly for 6 weeks. Two days after the final injection, donor-derived (CD45.2+) BM LSK cells were gated for analysis of the SLAM (LSKCD150+CD48−) cell population. For serial BMT, 2 million BM cells from the primary recipient mice were transplanted into sublethally irradiated secondary CD45.1+ recipient mice. Survival of the recipients was monitored and plotted by the Kaplan-Meier curve method.
Cell-cycle and apoptosis analysis
To analyze the cell cycle status of the HSC subsets, bone marrow cells were initially stained with antibodies against Lin+ cells, C-KIT, SCA-1, CD150 and CD48 described above. After incubation with these cell surface antibodies, the cells underwent fixation and permeabilization with transcription factor buffer set (BD Biosciences, #562725) according to the manufacturer’s instruction. After fixation, cells were incubated with APC-anti-Ki67 (BD Biosciences, #558615), washed and stained with PI. Cells were analyzed by flow cytometry. For the apoptosis detection, bone marrow cells were stained with the antibodies for the HSC surface markers, and then stained with APC-Annexin V (BD Biosciences, #550474) and 7 AAD. Annexin V-positive populations were determined as apoptotic cells using the FACS LSR II (BD Biosciences).
Statistical analysis
Student’s t-test was performed using GraphPad Prism v8 (GrapPad software). Comparaison of more than 2 groups was analyzed by one-way Anova test. Values of p<0.05 were considered statistically significant. Results are presented as mean±SD. * indicates p<0.05; **=p<0.01.
Go to :
Results and Discussion
We previously reported that chronic exposure of Fanca−/− mice to DNA crosslinking drug mitomycin C (MMC) induced the expansion of pre-leukemic HSCs (17). We have also shown that inactivation of Parp1 in Fanca−/− HSPCs caused hyper-active NHEJ, which was required for the predisposition of Fanca−/− HSPCs to leukemia in secondary recipients (18). To determine if pre-leukemic Fanca−/− HSC expansion required the NHEJ pathway, we crossed the Fanca−/− mice with a strain carrying the knockin DNA-PKcs3A/3A mutation, which selectively inactivates the NHEJ activity but does not affect the kinase activity of DNA-PKcs (15). To assess exclusively the response of HSCs without the effect of different BM microenvironment, we transplanted fetal LSK cells isolated from WT, Fanca−/−, DNA-PKcs3A/3A and DNA-PKcs3A/3A Fanca−/− embryos at E14.5 into lethally irradiated BoyJ mice. The recipients were exposed to DNA damage by intraperitoneally (ip) injection of a low dose of MMC (0.3 mg/kg; 15) weekly for 6 weeks. The recipient mice were sacrificed at the end of the treatment and analyzed for the frequency of donor-derived HSCs (CD45.2+LSKCD150+ CD48−; SLAM). As shown in Fig. 1A, a significant expansion of HSCs was observed only in the recipients transplanted with the Fanca−/− LSK cells (Fig. 1A). In contrast, inactivation of the NHEJ activity of DNA-PKcs prevented MMC-induced expansion of the Fanca−/− HSCs, as HSC expansion was not observed in recipients transplanted with DNA-PKcs3A/3AFanca−/− LSK cells (Fig. 1A). These results suggest that the NHEJ activity of DNA-PKcs is required for DNA damage-induced expansion of Fanca−/− HSCs.
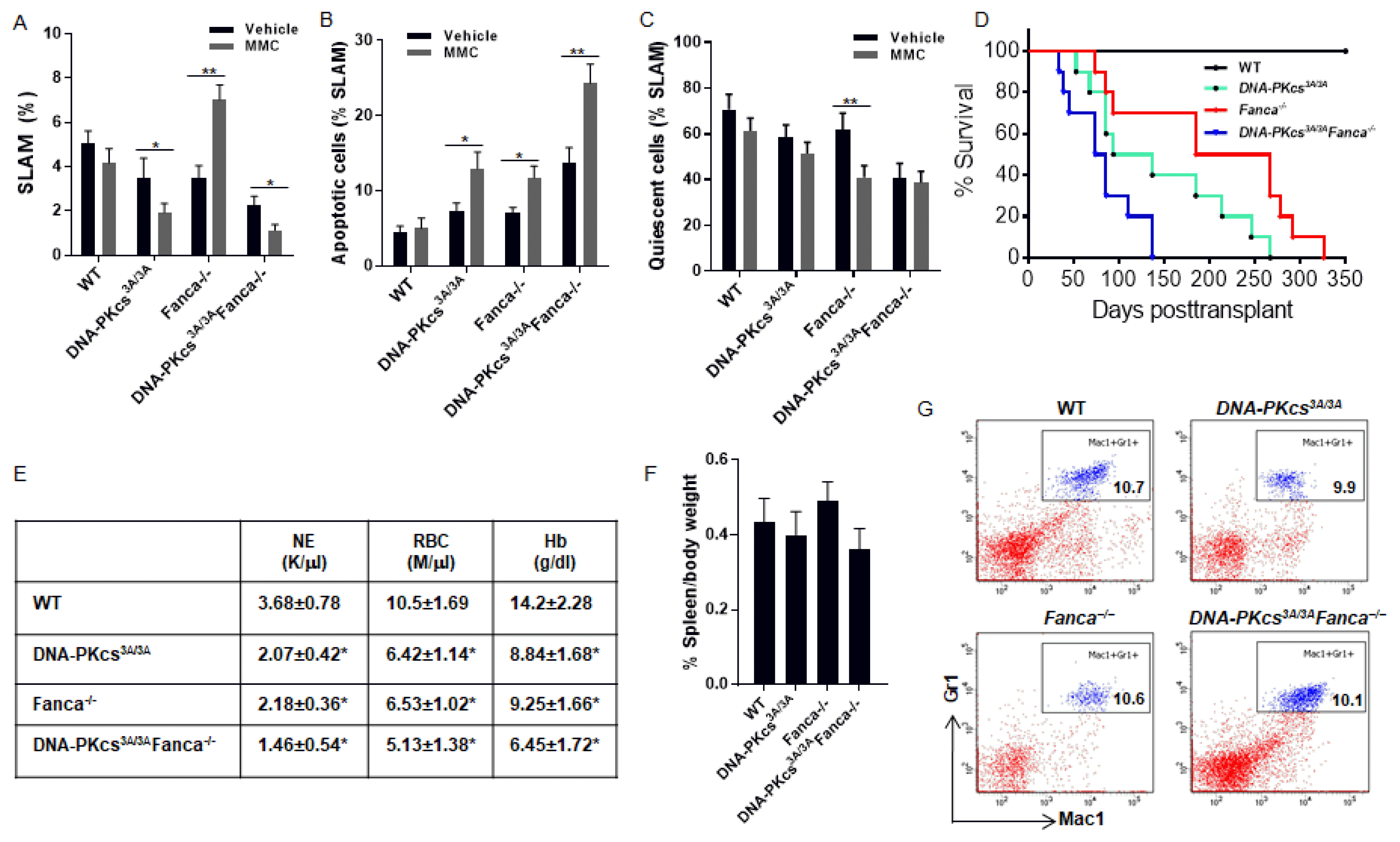 | Fig. 1Inactivation of the NHEJ activity of DNA-PKcs prevents the expansion of pre-leukemic Fanca−/− HSCs. (A) Inactivation of the NHEJ activity of DNA-PKcs prevents MMC-induced expansion of the Fanca−/− HSCs. 2,000 LSK cells from E14.5 embryos with the indicated genotype, along with 200,000 c-Kit-depleted protector cells, were transplanted into lethally irradiated BoyJ (CD45.1+) recipients. The recipients were i.p. injected with low dose of MMC (0.3 mg/kg) weekly for 6 weeks. Two days after the final injection, donor-derived (CD45.2+) BM LSK cells were gated for analysis of the SLAM (LSKCD150+CD48−) cell population. (B) MMC induces a significant increase in apoptosis in DNA-PKcs3A/3A, Fanca−/− and DNA-PKcs3A/3AFanca−/− SLAM cells. Donor-derived (CD45.2+) BM LSK cells were gated for SLAM population and analyzed for apoptosis by Annexin V and 7AAD. (C) MMC causes increased cell cycling in Fanca−/− HSCs. Donor-derived (CD45.2+) BM LSK cells were gated for the SLAM population and analyzed for the cell cycle using Hochest 33342/Ki67 staining. (D) Survival of transplant recipients. 2,000 LSK cells from E14.5 embryos with the indicated genotype, along with 200,000 c-Kit-depleted protector cells, were transplanted into lethally irradiated BoyJ recipients (n=10 per group). MMC (0.3 mg/kg) was then administrated to the recipients weekly for 6 weeks. Survival of the recipients plotted by the Kaplan-Meier curve method and analyzed by the log-rank test. (E~G) Anemia but no leukemia developed in primary recipient mice. The moribund mice described in (D) were subjected to analysis for peripheral blood count (E), and splenomegaly (F) and infiltration of mature myeloid cells (E) in the spleens. *p<0.05; **p<0.01.
|
To determine if the abrogation of MMC-induced Fanca−/− HSC expansion by NHEJ inactivation was associated with the status of apoptosis or quiescence, we analyzed apoptosis and cell cycle in the donor SLAM cell population. We found that MMC caused a significant increase in apoptosis in all three tested groups (DNA-PKcs3A/3A, Fanca−/− and DNA-PKcs3A/3AFanca−/−) of SLAM cells compared to the WT control (Fig. 1B), suggesting that the abrogation of Fanca−/− HSC expansion may be caused by increased apoptosis. However, MMC treatment induced significantly further reduction of quiescence in donor Fanca−/− SLAM cells but not in DNA-PKcs3A/3A or DNA-PKcs3A/3AFanca−/− cells (Fig. 1C). Thus, it appears that increased cycling may play a role in MMC-induced expansion of Fanca−/− HSCs.
To determine the biological consequence of DNA damage-induced Fanca−/− HSC expansion in the context of the NHEJ activity of DNA-PKcs, we performed serial BM transplantation and monitored the transplanted mice for leukemia development. The primary recipients transplanted with DNA-PKcs3A/3AFanca−/− donor cells succumbed to BM failure in less than 150 days post-transplant (Fig. 1D). All primary recipients receiving DNA-PKcs3A/3A or Fanca−/− donor cells also died from BM failure, albeit less severe than those receiving DNA-PKcs3A/3AFanca−/− donor cells (Fig. 1D). Analysis of the moribund mice showed peripheral neutropenia and anemia without signs of leukemia, as evidenced by the absence of splenomegaly and infiltration of mature myeloid cells in the spleens (Fig. 1E~G). However, we observed that recipients of Fanca−/− cells exhibited BM hypercellularity (Fig. 2A), and increased total donor-derived (CD45.2+) cells (Fig. 2B) and accumulation of donor SLAM cells (Fig. 2C) in the BM, compared to those of WT, DNA-PKcs3A/3A or DNA-PKcs3A/3AFanca−/− donor cells. Consistently, the secondary recipients of Fanca−/− cohorts gave rise to lethal leukemias within 52 weeks (Fig. 2D). The Fanca−/− leukemic mice showed infiltration of mature myeloid cells (Fig. 2E) and immature myeloid blasts (Fig. 2F) in the spleen. Consistent with these, we observed increased population of immature myeloid blasts in the peripheral blood smear from secondary recipient mice transplanted with Fanca−/− donor cells (Fig. 2G). Thus, these results indicate that the expanded Fanca−/− HSC compartment contains pre-leukemic stem cells that required the NHEJ activity of DNA-PKcs to induce leukemia in the secondary recipients.
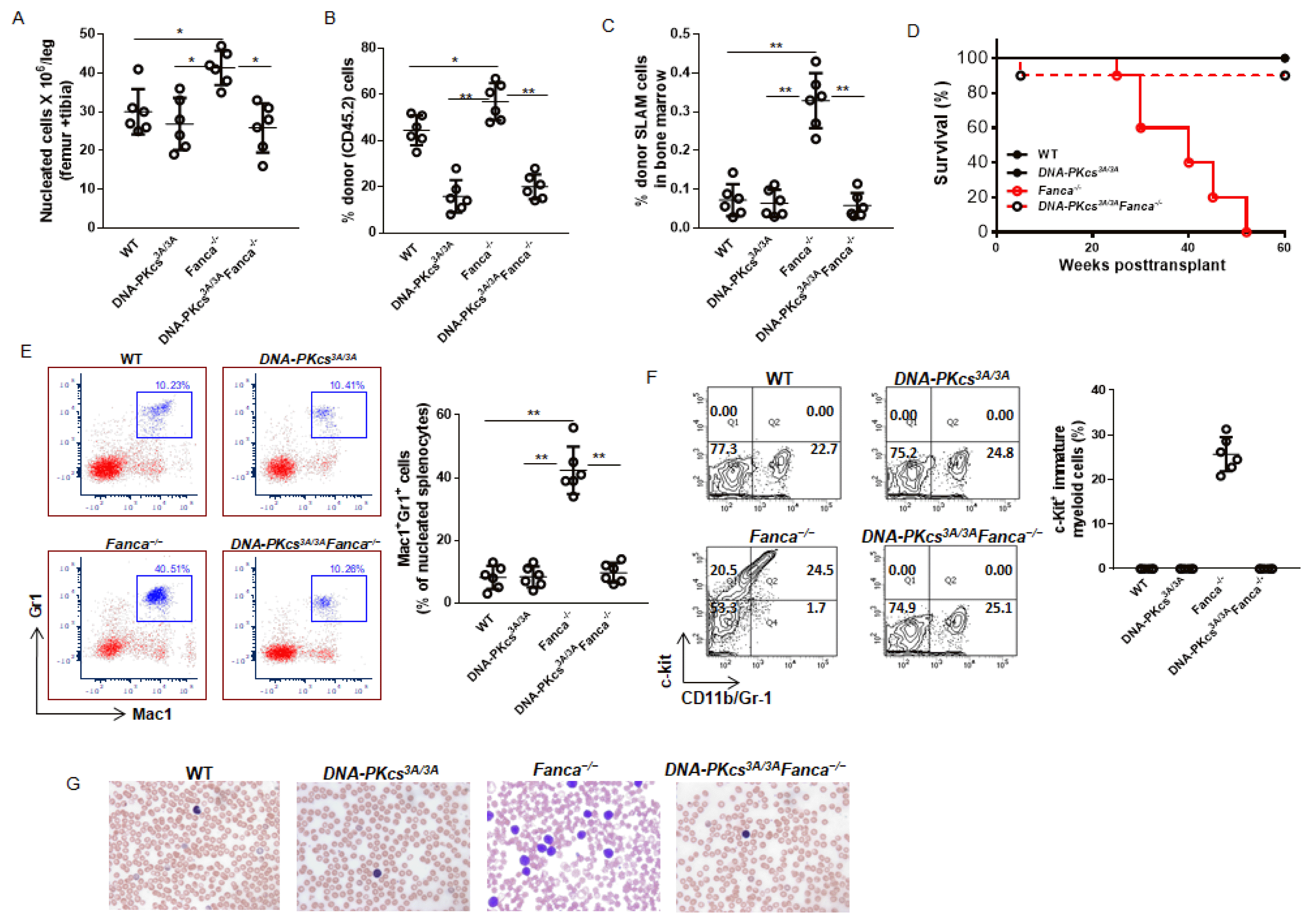 | Fig. 2The NHEJ activity of DNA-PKcs is required for the pre-leukemic Fanca−/− HSCs to induce leukemia in the secondary recipients. (A~C) Inactivation of the NHEJ activity of DNA-PKcs restores normal levels of BM cellularity and phenotypic HSCs in the BM of the secondary recipients. 2,000 LSK cells from E14.5 embryos with the indicated genotype, along with 200,000 c-Kit-depleted protector cells, were transplanted into lethally irradiated BoyJ (CD45.1+) recipients. The recipients were i.p. injected with low dose of MMC (0.3 mg/kg) weekly for 6 weeks. Two days after the final injection, the mice were sacrificed and analyzed for BM cellularity (A), total donor (CD45.2) engraftment (B), and donor-derived SLAM (LSKCD150+CD48−) cells (C). (D) Fanca−/− secondary recipients develop lethal leukemia. 2 million BM cells from the primary recipient mice described in (A) were transplanted into sublethally irradiated secondary CD45.1+ recipient mice. Survival of the recipients was monitored and plotted by the Kaplan-Meier curve method. n=8~10 mice for each group. (E, F) Increased myeloid infiltration in the spleen of the leukemic mice. Splenocytes from the recipient mice described in (D) were subjected to flow cytometry analysis for mature myeloid cells (E) and immature myeloid blasts (F). (G) Photomicrographs of Wright–Giemsa stain show increased blast cells in the peripheral blood smear from secondary recipient mice transplanted with Fanca−/− donor cells. *p<0.05; **p<0.01.
|
In this report, we employed an in vivo system, in which we specifically inactivated the NHEJ activity of DNA-PKcs in the Fanca−/− HSCs, to demonstrate that the NHEJ activity of DNA-PKcs is required for the expansion of pre-leukemic Fanca−/− HSCs. Our results suggest that the NHEJ pathway may functionally collaborate with FA deficiency in the process of leukemic transformation. We previously showed that loss of DNA damage response (DDR), due to FA deficiency, is correlated with the emergence and expansion of pre-leukemic Fanca−/− HSCs that give rise to leukemia in secondary transplanted recipients (17). However, how the multiple pathways (that is, FA, HR, NHEJ and other DDR pathways) cooperate within the DDR network in guarding against the initiation and progression of HSCs into leukemic transformation is not clear. Both FA and NHEJ pathways are involved in the repair of double-strand breaks (DSBs). Since the FA pathway can act to suppress the error-prone NHEJ pathway in DSB repair (11–14), it is conceivable that FA-deficient HSCs might preferentially use error-prone pathway for DSB repair, which may lead to high risk of pathological transformation. In this context, our finding is in line with the notion that impaired DDR due to FA deficiency and subsequently improper DSB repair as consequence of hyperactive NHEJ, resulting in genetic instability, may ultimately lead to leukemic transformation.
Go to :
 XML Download
XML Download