

 PDF
PDF Citation
Citation Print
Print


Introduction
Cancer is caused by environmental risk factors as well as genetic and epigenetic alterations, leading to uncontrolled cell proliferation and tumor formation (1). To explore the underlying mechanism of cancers, many researchers have attempted to investigate the cause of cancer (2–4).
In 2007, it was reported that overexpression of Yamanaka’s cocktails (OCT4, SOX2, KLF4, and C-MYC) could lead to reprogramming of differentiated cells into undifferentiated cells or induced pluripotent stem cells (iPSCs) (5, 6). iPSC technology in cancer research might help understanding the causes of cancer initiation and progression in vitro (7, 8). Furthermore, by reprogramming differentiated cells into an undifferentiated state based on iPSC technology, cancer cells might be reverted to their original state.
The normalization of cancer cells remains an important challenge due to the alteration of epigenetic modifications and expression of cancer-related genes (9). Understanding of the reprogramming of cancer cells would provide powerful tools for investigating the dynamic changes in the epigenetic and gene expression states of cancer cells.
Several groups have reported the reprogramming of cancer cells including melanoma, pancreatic cancer, gastrointestinal cancer, bladder cancer, lung carcinoma, and breast cancer (10–15). The cancer-derived iPSCs were distinct from parental cancer cells in their acquired sensitivity to chemotherapeutic agents and in tumorigenic activities (11, 13, 15–17). In another study, the authors hypothesized that human cancer cells could be converted into iPSCs and then differentiated again into cancer cells, which might be at an early developmental stage (18). This approach could provide a human model to study the early stages of cancer. However, reprogramming of cancer cells is less efficient and more time-consuming than that of normal somatic cells (19). The features important for their pluripotency, including morphology, gene expression, clonal expansion, immunocytochemistry, and teratoma formation, are not fully characterized (12, 15, 17, 20, 21).
Recent study implies that induction of pluripotency from malignant cancer cells was challenging compared to benign cancer cells (22). However, the exact reason for the differences regarding to reprogramming between malignant and benign cancer cells is unknown. To find out the exact mechanism that causes this difference might help understand malignancy in tumors. In general, primary cancer cells sourced from tumor tissues allow cancer study in in vivo like fashion. However, primary cancer cells isolated from tumor tissues are heterogeneous due to mixture of transformed cancer cells, supportive cells and tumor-infiltrating cells, which makes difficult to compare experimental results from the primary cells. Therefore, homogenous cancer cell lines would be beneficial for studying differences between malignant and benign cancer cells. Malignant breast cancer (MCF7) and benign breast cancer (MCF10A) cell lines have been used to identify the properties of malignant and benign cancer as in vitro model (4). Therefore, these cell lines are useful not only to verify the possibility of cancer reprogramming but also to compare difference between malignant and benign cancer cells regarding to reprogramming.
In this study, we attempted to reprogram normal human fibroblasts (BJ), MCF7, and MCF10A using the transcription factors OCT4, SOX2, KLF4, and C-MYC (OSKM). We compare transcriptome between BJ and cancer cells for finding the roadblock during cancer cell reprogramming. As a result, we identified the candidate barrier genes and their target genes during reprogramming.
Materials and Methods
Cell culture
BJ and PLAT-A cells were purchased from American Type Culture Collection (ATCC, VA, USA). Mouse embryonic fibroblasts (MEFs) were derived from C57BL6/N (KOATECH, Pyeongtaek, Korea) mouse strain embryos at embryonic day 13.5 after removing the head and all internal organs. MCF7 and MCF10A were obtained from Seoul National University, Korea, and Dongduk Women’s University, Korea. MEFs, BJ, and MCF7 were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Welgene, Gyeongsan-si, Korea) supplemented with 10% fetal bovine serum (Corning, NY, USA), 0.1 mM non-essential amino acids (NEAA; Gibco, MA, USA), and 1% penicillin/streptomycin (P/S; Welgene). MCF10A cells were cultured in MEGM kit medium (ATCC, CC-3150).
Retrovirus production
The pMX reprogramming vectors were purchased from Addgene (Cat. 17964, 17965, 17966, and 17967, respectively). The retroviral vectors were produced by transfection of these vectors (15 μg each) into PLAT-A cells in 150 μl of iN-fect in vitro transfection reagent (iNtRON, Seongnam, Korea). The first virus-containing supernatant of the transfected cells was collected 24 h after transfection and replaced with fresh medium, which was collected 24 h later as the second virus-containing supernatant. Each virus-containing supernatant was filtered through a 0.45 μm syringe membrane filter. The pMX-DsRed vector was used to evaluate viral infection efficiency.
iPSC reprogramming
BJ, MCF7, and MCF10A cells seeded in a 6-well plate (SPL Life Science, Pocheon, Korea) at 2×105 cells per well were incubated at 37°C with equal volumes of the virus-containing supernatants supplemented with 4 μg/ml polybrene (Sigma-Aldrich, MO, USA). After 24 h, the first virus-containing supernatant was replaced with the second one. Four days after the first-infection, the cells were passaged by using 0.25% trypsin (Gibco) and plated on MEF feeders. After 24 h, the medium was changed to iPSC generation medium, consisting of Knockout DMEM (Gibco) supplemented with 20% Knockout Serum Replacement (Gibco), 0.1 mM NEAA, 1% P/S, 1% 2-mercaptoethanol (Gibco), 10 ng/ml of basic fibroblast growth factor (Peprotech, Seoul, Korea), 0.5 mM sodium butyrate (Sigma-Aldrich), 2 μM SB431542 (Tocris Bioscience, BR, UK), and 0.5 μM PD0325901 (Sigma-Aldrich). The medium was changed every day. Within 21 days post-infection, BJ-iPSCs expanded to a size suitable for transfer. BJ-iPSC colonies were mechanically transferred onto Matrigel (Corning)-coated 4-well dishes by using a 10 μl pipette tip and cultured in mTeSR medium (Stemcell Technologies, Vancouver, Canada).
RT-PCR and quantitative PCR
Total RNA was purified using the RNeasy Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions; 1 μg of total RNA was converted into cDNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, CA, USA) according to the manufacturer’s instructions. RT-PCR was performed with Ex Taq Polymerase (TaKaRa, Shiga, Japan) according to the manufacturer’s instructions. Quantitative PCR (qPCR) was performed with the TaqMan PCR Master Mix (Thermo Fisher Scientific, MA, USA) and SYBR Green (Enzynomics, Daejeon, Korea) on a LightCycler 1536 qPCR system (Roche, BA, Switzerland). Expression levels were normalized to that of GAPDH. qPCR was performed with gene-specific primers (Supplementary Table S1).
Alkaline phosphatase staining
Cells were fixed in 4% paraformaldehyde for 15 min at room temperature and stained for alkaline phosphatase (AP) with the Leukocyte Alkaline Phosphatase kit (Stemgent, MA, USA) following the manufacturer’s instructions.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 20 min at room temperature and washed 3 times with Dulbecco’s Phosphate Buffered Saline (DPBS; Hyclone Laboratories, UT, USA). The cells were then permeabilized and blocked with DPBS containing 0.5% Triton X-100 (Sigma-Aldrich) and 1% bovine serum albumin (Sigma-Aldrich) for 1 h at room temperature. Cells were incubated with primary antibodies (Pluripotency Antibody kit, Cell signaling, MA, USA) overnight at 4°C, washed 3 times with DPBS, and incubated with secondary antibodies (against mouse IgG, mouse IgM, or rabbit IgG; all from Cell signaling) for 1 h at room temperature in the dark. Nuclei were stained with Hoechst33342 (Invitrogen, CA, USA).
RNA-sequencing analysis
The amount and quality of total RNA were assessed using an Agilent 2100 Bioanalyzer RNA kit (Agilent Technologies, CA, USA). Total RNA was used to prepare mRNA sequencing libraries with the Illumina TruSeq Stranded mRNA Sample Preparation kit (Illumina, CA, USA) according to manufacturer’s instructions. We used the Agilent 2100 Bioanalyzer DNA kit (Agilent Technologies) to evaluate the quality and size of the libraries. All libraries were quantified with qPCR using the CFX96 Real Time System (Bio-Rad, CA, USA) and sequenced with a paired-end 75-bp run plus a single 8-bp index read run on a NextSeq 500 sequencer (Illumina).
Preprocessing and genome mapping
Potential sequencing adapters and low-quality bases in the raw reads were trimmed by Skewer (23). The options –x AGATCGGAAGAGCACACGTCTGAACTCCAGTCA and –y AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT were used for the common sequence of the Illumina TruSeq adapters and the option -q 0 -l 25 -k 3 -r 0.1 -d 0.1 was used for trimming low-quality 5′ and 3′ ends of the raw reads. The trimmed high-quality reads were mapped to the human reference genome by STAR software (24). A strand-specific sequencing library was prepared according to the protocol in the Illumina’s strand-specific library preparation kit; the strand-specific library option, --library-type=fr-firststrand was used for mapping.
Quantifying gene expression and differential gene expression analysis
To convert the numbers of mapped reads into the gene expression values, the Cufflinks package (25) with the strand-specific library option, --library-type=fr-firststrand and other default options was used. The gene annotation of the human reference genome mm10 was accessed through the UCSC genomes browser (https://genome.ucsc.edu) and used in GTF format, and the expression values were calculated as fragments per kilobase of transcript per million fragments mapped. The Cuffdiff software in Cufflinks was used to analyze differentially expressed genes between the two selected biological conditions (26). To compare the expression profiles between samples, the normalized expression values of the selected few hundred differentially expressed genes were clustered by unsupervised in-house R scripts.
Functional category and network analysis
Overlap between the analyzed differentially expressed genes and functionally categorized genes, including biological processes of Gene ontology (GO) and KEGG pathways, was tested by using g:Profiler (http://compbio.mit.edu/cummeRbund/). Cytoscape was used to identify gene interaction network and to visualize networks constructed using selected candidate barrier genes and their target genes as nodes (27).
Results
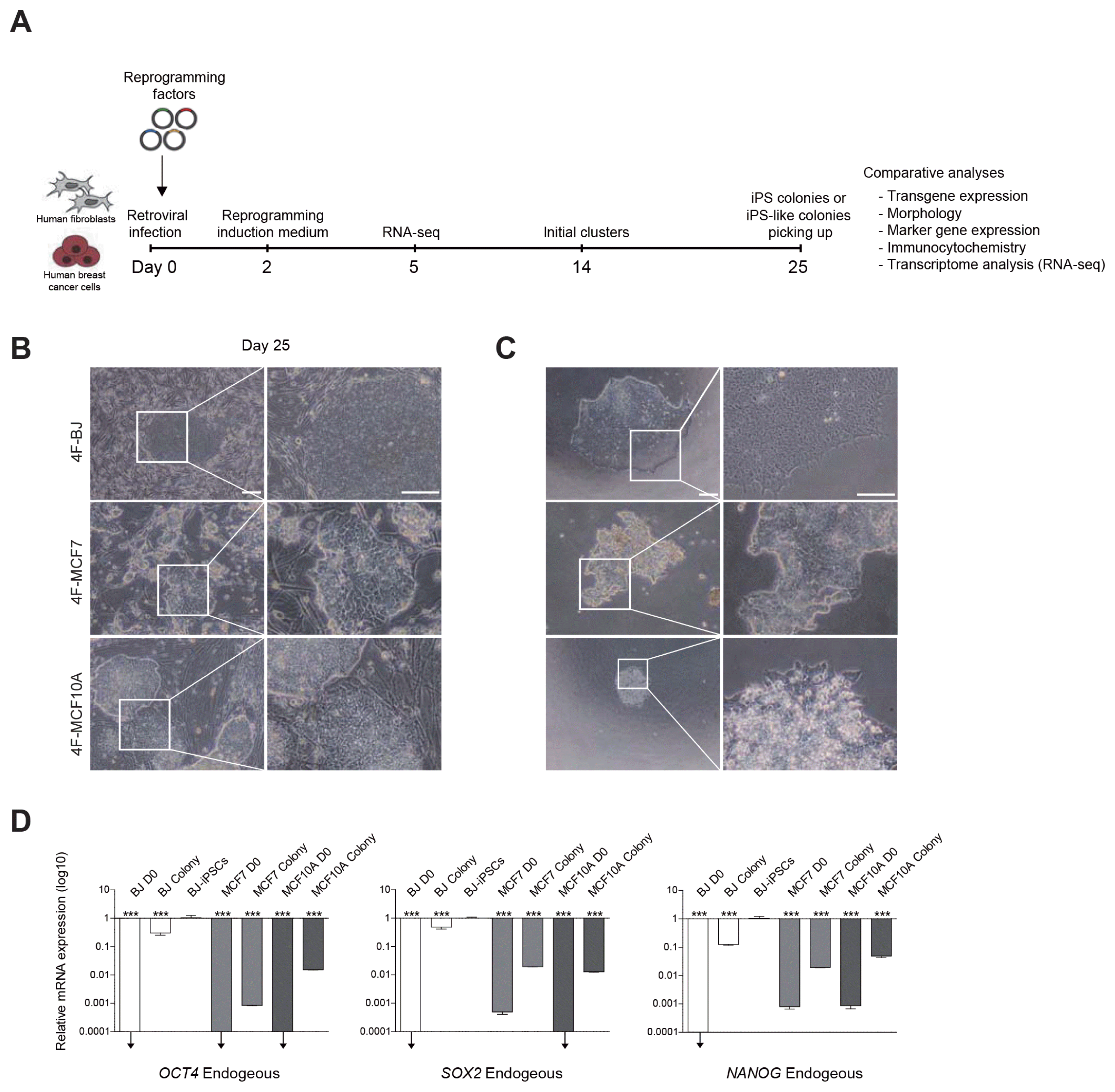
Generation and characterization of iPSCs from human fibroblasts and cancer cells
To test whether the retroviral vector system could efficiently deliver OSKM into cancer cells, we first infected MCF7 and MCF10A cells with a retroviral vector encoding a fluorescent protein, DsRed. Infection efficiency was approximately 80%~90%, which was similar to that of BJ (Supplementary Fig. S1B). To generate iPSCs, we infected BJ, MCF7 and MCF10A cells with retroviruses encoding OSKM (Fig. 1A). After 25 days, we observed iPSC-like colonies forms from all three cell lines (Fig. 1B). For further expansion and establishing clonal lines, an individual colony of each group was picked up and plated onto Matrigel-coated dishes. The colony from OSKM-transduced BJ (4F-BJ) could be stably maintained and established an iPSC line. In contrast, we were unable to maintain and expand the colonies from OSKM-transduced MCF7 (4F-MCF7) and MCF10A (4F-MCF10A) (Fig. 1C). We next investigated the cellular properties of these iPSC-like colonies derived from MCF7 and MCF10A.
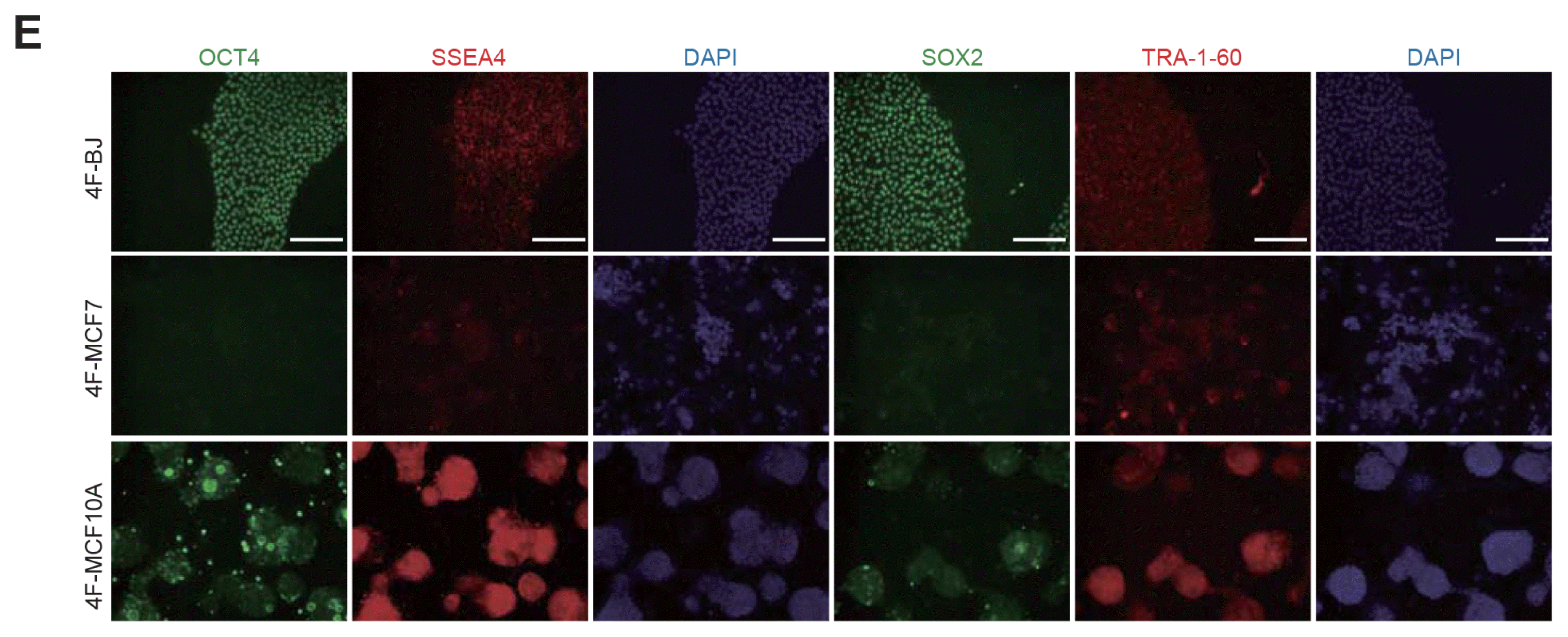
We performed qPCR to compare the expression levels of endogenous pluripotency markers in BJ, MCF7, and MCF10A iPSC-like colonies at day 25. BJ-iPSCs were used as a positive control. The representative of pluripotency genes, OCT4, SOX2, and NANOG, were activated in BJ colonies, but not in MCF7 and MCF10A colonies (Fig. 1D). In general, these genes are gradually activated during reprogramming (28). To further check the pluripotent state of iPSC-like colonies, we performed AP-staining analysis of iPSC-like colonies from BJ, MCF7, and MCF10A. We detected AP-positive colonies derived from BJ but not MCF7 and MCF10A (Supplementary Fig. S1A). The morphological features and non-specific AP-positive staining patterns of iPSC-like colonies from MCF7 and MCF10A differed from those of BJ. We also confirmed the induction of pluripotency at the protein level. The pluripotency markers OCT4, SOX2, and NANOG were observed in a BJ iPSC-like colony, but not in MCF7 and MCF10A iPSC-like colonies at day 25, as determined by immunostaining (Fig. 1E). Although we could detect dim signals of these markers in MCF10A iPSC-like colony, it was not exclusively located in the nucleus. Interestingly, the pluripotency surface markers SSEA4, TRA-1-60, and TRA-1-81 were expressed in all iPSC-like colonies from BJ, MCF7, and MCF10A (Fig. 1E, Supplementary Fig. S1C). This indicates that SSEA4, TRA-1-60, and TRA-1-81 are not sufficient for identifying fully reprogrammed cells derived from cancer cells.
These data suggest that MCF7 and MCF10A show relatively poor induction of pluripotency due to the lack of expression the core pluripotency genes such as OCT4, SOX2, and NANOG.
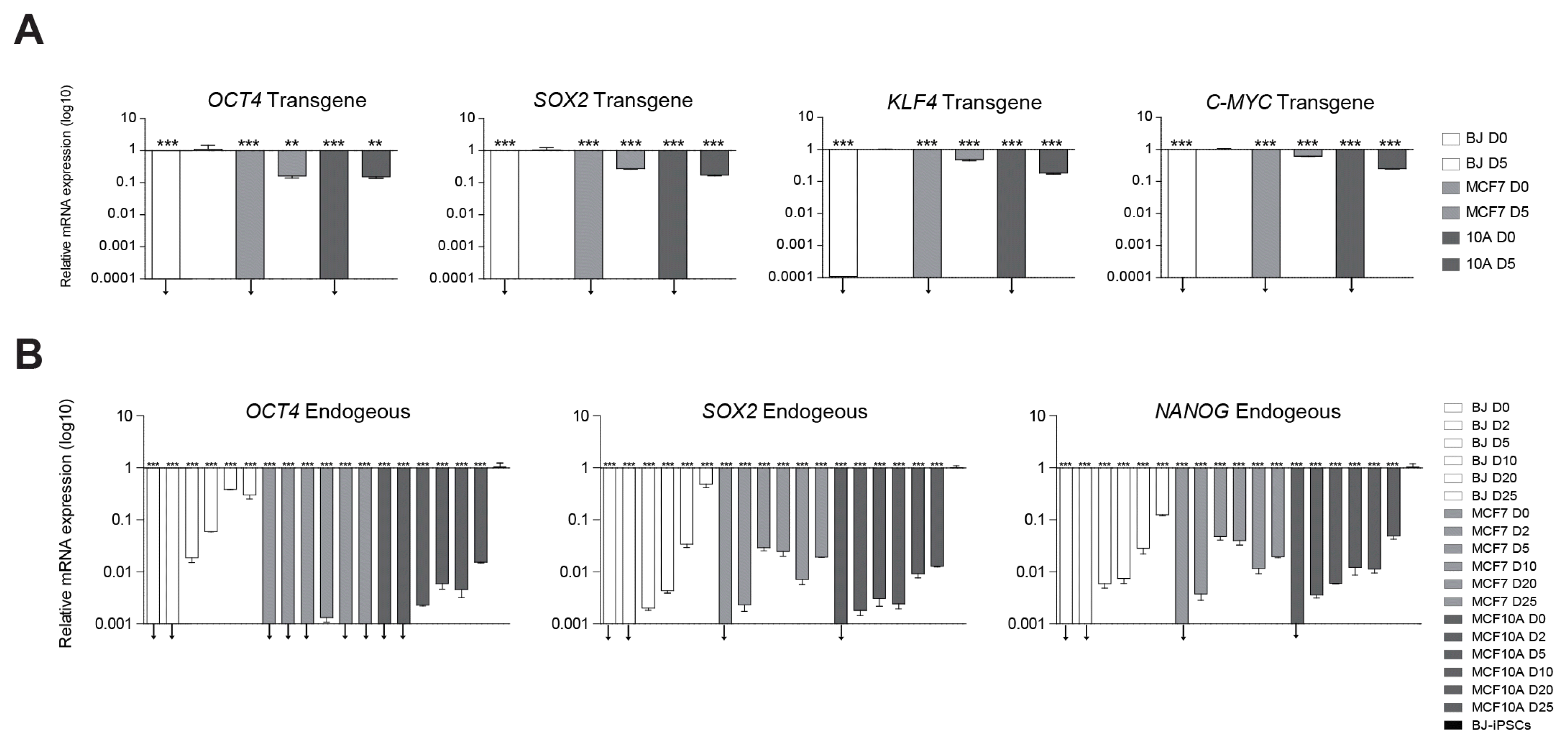
Comparison of gene expression in human fibroblasts and cancer cells during reprogramming
Although previous studies described the reprogramming kinetics in human fibroblasts in detail, that of cancer cells remains unknown (29). To elucidate the gene expression patterns during reprogramming, we followed the time-course of gene expression by qPCR. We first checked the transduction efficiency by evaluating the induction level of transgenes by qPCR and found it to be similar in BJ, MCF7, and MCF10A at day 5 after transduction (Fig. 2A). These data indicate that our retroviral system efficiently delivers the reprogramming factors into all three types of cells. Next, we checked the endogenous expression levels of pluripotency genes, OCT4, SOX2, and NANOG at days 0, 2, 5, 10, 20, and 25. As expected, the expression of the pluripotency genes began to gradually increase from day 0 in BJ, whereas these genes were not up-regulated in MCF7 and MCF10A during reprogramming (Fig. 2B).
Our data revealed that the endogenous OCT4, SOX2, and NANOG were properly up-regulated in BJ, whereas endogenous OCT4, SOX2, and NANOG failed to be up-regulated in MCF7 and MCF10A compared to BJ, despite successful transduction of the reprogramming factors.
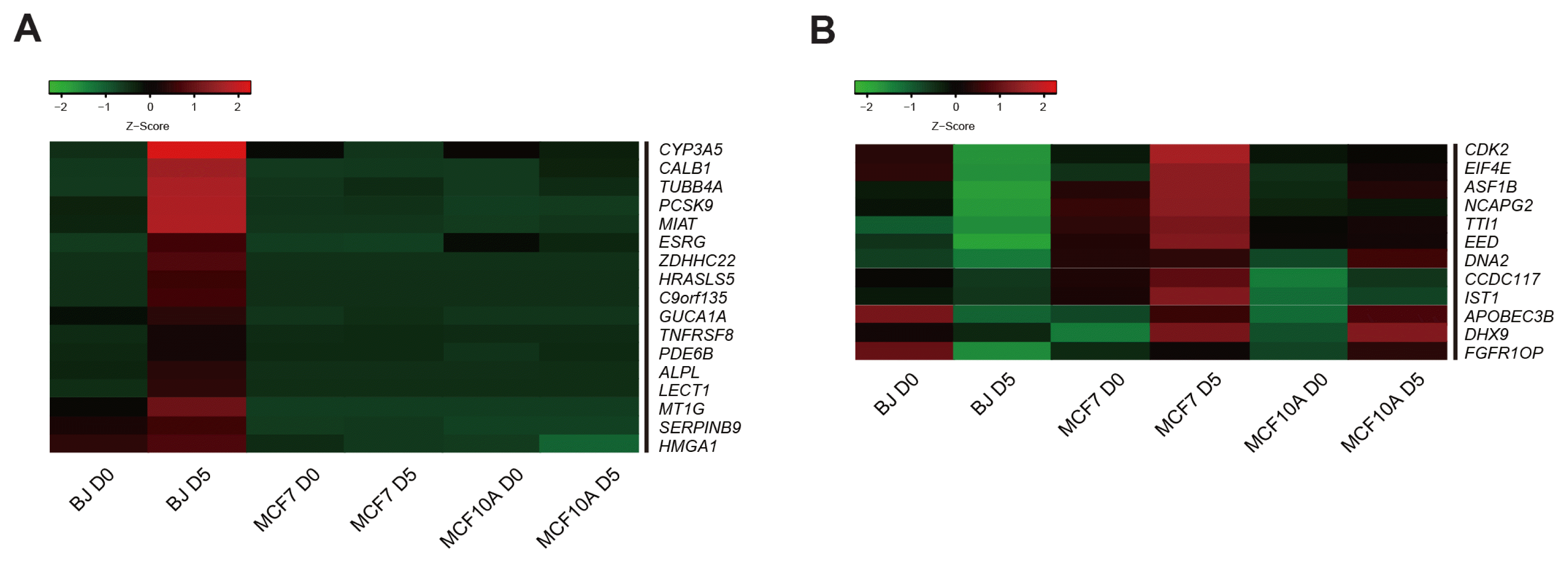
Molecular characterization of human fibroblasts and cancer cells at day 5 of reprogramming stage
Unexpectedly, we could not induce pluripotency in 4F-MCF7 and 4F-MCF10A by using reprogramming factors. To reveal the mechanism that limits reprogramming in cancer cells, we performed RNA-seq analysis in BJ, MCF7, and MCF10A during reprogramming. To this end, we selected the genes, which are essential for the early stage of reprogramming (29). Among them, using heatmap analysis, we found 17 candidate barrier genes that are up-regulated in BJ D5, but not in MCF7 D5 or MCF10A D5 (Fig. 3A). Furthermore, we identified 12 candidate barrier genes that are down-regulated in BJ D5 but not in MCF7 D5 or MCF10A D5 by heatmap analysis (Fig. 3B).
These data suggest that a total of 29 candidate barrier genes (17 genes, which are up-regulated gene in BJ, but not in MCF7 and MCF10A and 12 genes, which are down-regulated genes in BJ, but not in MCF7 and MCF10A) might act as a roadblock to achieve pluripotency in an early reprogramming stage in cancer cells.
Genetic interaction network of candidate barrier genes and their target genes at an early stage of reprogramming
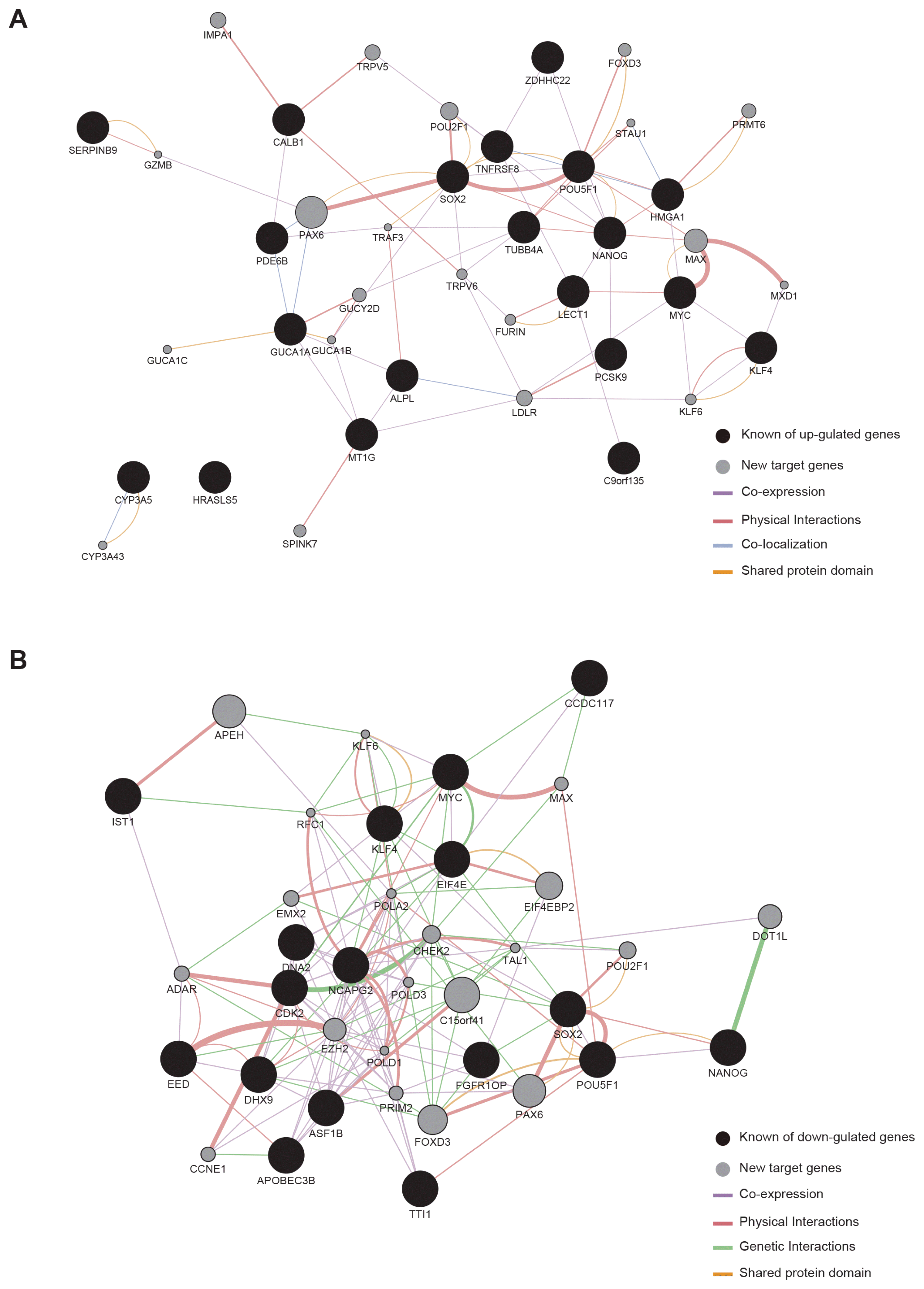
To gain a deeper insight into the mechanism of action of the candidate barrier genes, we constructed a genetic interaction network (GIN) based on their target genes as well as candidate barrier genes. First of all, we tried to identify the target genes of 29 candidate barrier genes we found. Based on the RNA-seq data, we identified 40 target genes using Cytoscape software and constructed a GIN for investigating interaction between candidate barrier genes and their target genes.
Among a total 40 putative target genes identified, 20 genes (PAX6, MAX, POU2F1, IMPA1, LDLR, TRPV5, PRMT6, GUCY2D, FOXD3, SPINK7, FURIN, KLF6, TRPV6, CYP3A43, GUCA1C, GUCA1B, STAU1, MXD1, TRAF3, and GZMB) were up-regulated by the 17 genes that were up-regulated in BJ, but not in MCF7 or MCF10A (Fig. 4A). In GO analysis, a number of genes were associated with co-expression, physical interactions, co-localization, and shared protein domain. The other 20 target genes (C15orf41, APEH, PAX6, FOXD3, EIF4-EBP2, DOT1L, EZH2, CHEK2, POU2F1, EMX2, ADAR, CCNE1, PRIM2, MAX, POLD3, TAL1, POLA2, POLD1, RFC1, and KLF6) were related to the 12 genes that were down-regulated in BJ but not in MCF7 or MCF10A (Fig. 4B). GO analysis showed that these genes were related to co-expression, physical interactions, genetic interactions, and shared protein domain.
Overall, these results suggest that the complex connection of barrier genes to their target genes could be the major obstacle for reprogramming.
Discussion
The iPSC technology is a powerful tool for transformation of the differentiated cells into the pluripotent cells (30). Many reports have been demonstrated that iPSCs can be generated by ectopic expression of the Yamanaka’s cocktail from various cell types such as human fibroblasts (5, 6), blood cells (31), keratinocytes (32), and melanocytes (33). Although previous studies attempted to generate iPSCs from breast cancer cell, MCF7 and MCF10A (15, 17), the reprogrammed cells were not fully characterized; they had unusual morphology, lack of pluripotency markers, and limited differentiation potential in vitro and in vivo. Moreover, another study suggested that cancer cells are resistant to cellular reprogramming (9). In our study, to reveal the underlying mechanism of the limited reprogramming capacity of breast cancer cells, we attempted to find the candidate barrier genes and their target genes by global transcriptome analysis. To this end, we first generated iPSCs from BJ, MCF7, and MCF10A cells by using retroviral vectors carrying OSKM. We found that genuine colonies were not formed from MCF7 and MCF10A. We next checked the expression levels of endogenous pluripotency genes in time-course manner. Interestingly, 4F-MCF10A showed relatively higher expression of endogenous mRNA and protein level rather than 4F-MCF7. When considering for reprogramming potential, MCF10A seems to be more likely to be reprogrammed than the MCF7, consistent with previous report (22).
OCT4, SOX2, and NANOG are considered as core factors for the transcriptional regulatory network that specifies pluripotent states because of their expression patterns and their key roles during the early development (34). As a result, endogenous expression levels of OCT4, SOX2, and NANOG were not up-regulated in MCF7 and MCF10A during reprogramming process. On the other hand, that of these genes was gradually increased in BJ. This result suggests that cancer cells display abnormal properties that most healthy cells do not possess such as genetic mutations and epigenetic alterations (9). Therefore, we speculate that this dysregulation in cancer cells would limit activation of the core transcriptional pluripotency circuitry.
To investigate transcriptome profile changes during reprogramming, we performed RNA-seq analysis in BJ, MCF7, and MCF10A at day 5 after transduction. By comparing their transcriptomes, we screened for candidate barrier genes involved in the early stage of reprogramming. We found 29 candidate barrier genes and categorized them into two groups: 17 candidate barrier genes that are not up-regulated in MCF7 and MCF10A but not in BJ at day 5. In addition, 12 candidate barrier genes, which are not down-regulated in MCF7 and MCF10A but not in BJ at day 5.
For a deeper understanding of the initial reprogramming process, we sought to identify the target genes of the 29 candidate barrier genes using Cytoscape, and found 40 target genes. There was a complex connection between the candidate barrier genes and target genes, and this complex connection may be strongly affected cancer cells as an important obstacle in the early stage of reprogramming. Our GO analysis of the 40 genes revealed significant enrichment of the co-expression, physical interaction, co-localization, genetic interactions, and shared protein domain. These complicated signaling pathways might be critical for cell survival and maintenance of cancer properties.
Our RNA-seq data (Supplementary Table S2 and S3) suggested that, among the target genes, PRMT6, MXD1, and EZH1 were highly expressed in cancer cells during reprogramming regardless of the ectopic expression of OSKM, indicating that these genes could be obstacles in the early stage of cancer cell reprogramming.
Previous studies demonstrated that PRMT6 (protein arginine N-methyltransferase) is highly expressed in cancer cells and is related to global DNA hypermethylation (35). The MXD1 gene interacts with the MAX gene and inhibits transcription of C-MYC-regulated genes (36). The histone-lysine N-methyltransferase EZH1 regulates a variety of genes and affects complex epigenetic regulation in cancer cells (37). We suggest that these three genes are one of the roadblock to obtain pluripotency in cancer cells.
The potential of iPSC technology in cancer research has an infinite possibility. However, it has been begun to be explored since few years ago. Our in vitro model of the breast cancer-derived iPSCs could be used as platforms to screen anti-cancer drug and to discover prognostic biomarkers. In addition, our data provides an integrative approach for identifying the complex connection between candidate barrier genes and their target genes that may act as an obstacle during reprogramming in cancer cells. Finally, understanding the mechanism of cancer cell reprogramming would be valuable to therapeutic application in cancer.
 XML Download
XML Download