

 PDF
PDF Citation
Citation Print
Print


Introduction
The bone marrow produces new blood cells from hematopoietic stem cells that are capable of self-renewal and can generate all blood lineages (1). Nonetheless, non-hematopoietic stem cells supply and constitute the supportive stromal structure to assist with hematopoiesis by differentiating into somatic cells of the mesenchymal lineage (2) and are collectively defined as marrow stromal cells or mesenchymal stem cells (MSCs) (3).
MSCs are isolated from a variety of adult tissues and can differentiate into mesenchymal as well as nonmesenchymal cell types including neurons (4). Although MSCs are inherently multipotent, their uncertain ability to undergo in vivo transdifferentiation limits their applications in regenerative medicine. Moreover, most of the injected MSCs are prone to clearance and only a few localize within injured tissues (5). In contrast, the ability of MSCs to modulate inflammatory or immune responses has aroused considerable interest in relevant clinical and translational studies. MSCs suppress the activation and proliferation of lymphocytes and modulate several subsets of immune cells including dendritic cells, natural killer cells, and macrophages (6, 7). The beneficial effects of MSCs in a broad range of inflammatory or autoimmune disorders have been reported, including graft-versus-host disease, colitis, acute pancreatitis, and atopic dermatitis (8–11). Presently, there is a consensus that the immunosuppressive influence of MSCs depends on secreted factors as well as direct cell contact. Various immunoregulatory factors such as indoleamine 2,3-dioxygenase, prostaglandin E2, hepatocyte growth factor, transforming growth factor β, heme oxygenase 1, interleukin (IL) 10, nitric oxide (NO), and tumor necrosis factor α (TNF-α)-stimulated gene 6 protein (TSG-6) have been implicated in MSC-mediated immunosuppression (12, 13). Nonetheless, the pivotal role of these factors in MSC-mediated immunomodulation is not completely elucidated because inhibition of any of these factors does not abrogate the immunosuppression elicited by MSCs (12). Moreover, whether a few MSCs at an injury site can produce sufficient paracrine factors to influence systemic or local inflammatory responses remains undetermined. Therefore, MSC-mediated immunosuppression might be based on currently unknown mechanisms.
To maintain the systemic control over inflammation and immune responses in an organism, signaling between the immune and neuroendocrine systems is indispensable during host defense (14). The interplay between these two systems constitutes a closely interconnected feedback network for control over inflammatory responses. Local acute inflammation is characterized by an early release of proinflammatory cytokines including TNF-α, interferon γ (IFN-γ), and ILs. They ensure host homeostasis by driving immediate cerebroneural activation followed by local immune, systemic neuroendocrine, and local neural responses (15, 16). Neuroendocrine signaling pathways including the hypothalamic–pituitary–adrenal axis and autonomic nervous system (ANS) act as anti-inflammatory regulators (16, 17). Acetylcholine (ACh), a primary parasympathetic neurotransmitter, is released by efferent vagus nerve (EVN) fibers in response to IL-1 secreted by activated innate-immunity cells; this action results in the negative feedback control of inflammation during an inflammatory response in the gut or peritoneum (14). ACh binds to nicotinic and muscarinic cholinergic receptors on immune cells (18, 19). In macrophages, the cholinergic inhibition of inflammation is driven specifically by nicotinic ACh receptor (nAChR) in which α7 is the major subunit (20, 21). Furthermore, cholinergic receptors have been identified on human lymphocytes (18, 19), and their activity correlates with T-cell activation (22). nAChR α7 has been suggested to participate in the release of various cytokines and in antibody production (23). Hoogduijn et al. (24) have detected the expression of choline acetyltransferase (ChAT) and acetylcholinesterase (AChE) and the presence of ACh in MSCs. Additionally, they found that MSCs express nAChR subunits α3, α5, and α7 and muscarinic AChR 2 (24).
Therefore, our aim was to determine the immunomodulatory mechanisms elicited by MSCs during inflammation. In our study, we demonstrate that human bone marrow–derived (hBM) MSCs inhibit the activation and proliferation of activated immune cells (AICs) under inflammatory conditions. We observed that the inflammatory milieu created by the immune-cell activation causes MSCs to manifest neuronlike characteristics and release ACh. This process in turn inhibits the inflammatory responses through nAChR and might act as a probable mechanism by which MSCs can suppress immune cells via the ACh–nAChR signaling pathway.
Materials and Methods
This work was approved by Inha Hospital Institutional Review Board (#14-082) and was performed in accordance with the provisions of the Declaration of Helsinki (as revised in Edinburgh, 2000). The donors signed informed consent forms. The animal experiments were approved by Inha University Animal Research Committee.
Characterization and culturing of MSCs
hBM MSCs were provided by SCM Lifescience Co., Ltd. (Incheon, South Korea). MSCs were isolated and characterized based on cell shape, marker expression, and mesenchymal differentiation. They were cultured at 37°C in a humidified CO2 incubator in Dulbecco’s modified Eagle’s medium (DMEM) with low glucose concentration (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 10% of fetal bovine serum (Gibco-BRL), 1% of a penicillin/streptomycin solution (Gibco-BRL), and 1% of MycoGone (Genlantis, San Diego, CA, USA). At 70~80% cell confluence, they were dissociated with 0.05% trypsin/EDTA and subcultured for further expansion (10).
An immunosuppression assay and inflammatory conditions
To simulate experimental inflammatory conditions, peripheral blood mononuclear cells (PBMCs) were isolated from two healthy donors, and 105 PBMCs from each donor were cocultured with 4×104 MSCs in a 96-well plate for 5 days to carry out a mixed lymphocyte reaction (MLR). To perform mitogen activation, 2×105 PBMCs were stimulated with 1 μg/mL phytohemagglutinin (PHA, a lectin) (Sigma, St. Louis, MO, USA) and cocultured with 4×104 MSCs for 3 days. The lymphocyte proliferation was determined by monitoring the incorporation of [3H]thymidine (1 μCi/well) during the final 12~16 h of incubation. The radioactivity emitted by the incorporated [3H]thymidine was measured on a β-counter (Perkin-Elmer, Waltham, MA, USA).
An enzyme-linked immunosorbent assay (ELISA)
A conditioned medium (CM) was harvested from the cultures of cells treated with either a nAChR antagonist or agonist in coculture experiments. The secreted proinflammatory cytokines were quantified using the ELISA kits for TNF-α (cat. # 555212; BD Biosciences) and IFN-γ (cat. # 555141; BD Biosciences). The CM from MLR- or PHA-activated PBMCs was assayed for the presence of nerve growth factor (NGF) (cat. # DY256-05; R&D Systems, Minneapolis, MN, USA) and brain-derived growth factor (BDNF) (cat. # DBD00; R&D Systems).
RNA isolation, semiquantitative reverse-transcription polymerase chain reaction (RT-PCR), and quantitative PCR (qPCR)
Total RNA was extracted by means of the easyBlue RNA isolation reagent (iNtRON, Sungnam, South Korea). cDNA was synthesized from 1 μg of total RNA using the AccuPower cDNA synthesis kit (Bioneer, Daejeon, South Korea). RT-PCR was carried out with the AccuPower PCR Premix (Bioneer). The amplicons were subjected to electrophoresis in 1% agarose gels containing SYBR Safe (Invitrogen, Carlsbad, CA, USA) and quantified on an LAS4000 mini fluorescence image analyzer (Fuji Photo Film, Tokyo, Japan). The PCR primer sequences are listed in Supplementary Table S1. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the internal control. Primers for qPCR were purchased from Applied Biosystems (Foster City, CA, USA). The primers were as follows: tropomyosin receptor kinase (Trk) genes TrkA (Hs01021011), TrkB (Hs00178811), and TrkC (Hs00176797), p75 neurotrophin receptor (p75NTR) (Hs00609977), nAChR α7 (Hs04189909), nAChR α5 (Hs00181248), ChAT (Hs00252848), and 18S rRNA (Hs03928985). The qPCR was conducted on a StepOne Real-Time PCR system (Applied Biosystems).
Flow cytometry
Anti–glial fibrillary acidic protein (GFAP; cat. # 51449), anti–class III β-tubulin (TUJ1; cat. # 560381), and antinestin (cat. # 56130; all BD Biosciences) antibodies were used to perform flow-cytometric analysis. Isotype-matched antibodies served as controls. Cells were analyzed on a FACSCalibur flow cytometer (BD Biosciences).
Western blot analysis
Cells were washed twice with phosphate-buffered saline (PBS) and disrupted for 30 min on ice in lysis buffer consisting of 50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA (Sigma), 1 mM sodium orthovanadate (Sigma), 1 mM sodium fluoride, 1 mM PMSF (Sigma), 1% Triton X-100, a protease inhibitor cocktail (Pierce, Rockford, IL, USA), and a phosphatase inhibitor cocktail (Pierce). Cell debris were removed by centrifugation at 15,000 ×g for 15 min, and the supernatants were collected into new micro-centrifuge tubes. Protein concentration was measured using the BCA Protein Assay Reagent Kit (Pierce). Equal amounts of protein were resolved by SDS polyacrylamide gel electrophoresis in 10% gels under reducing conditions and were electrotransferred onto Immobilon P membranes (EMD Millipore, Billerica, MA, USA). The following antibodies were employed for immunodetection: anti-TrkA (cat. # 06-574; EMD Millipore), anti-TrkC (cat. # 3376; Cell Signaling Technology, Danvers, MA, USA), anti-p75NTR (cat. # 8238; Cell Signaling Technology), anti-ChAT (cat. # AB144P; EMD Millipore), anti–nAChR α7 (cat. # ANC-007; Alomone, Jerusalem, Israel), and anti–β-actin (cat. # SC-47778; Santa Cruz Biotechnology, Dallas, TX, USA). After incubation with an appropriate secondary antibody conjugated to horseradish peroxidase, bands were detected by means of enhanced chemiluminescence reagent West-Zol Plus (iNtRON). β-Actin served as a loading control.
An ACh assay
MSCs were incubated with the CM obtained from cultures after MLR or PHA stimulation experiments. After 24 h, the medium was harvested. The level of ACh and choline in the harvested medium was quantified using the Enzychrom Acetylcholine Assay Kit (cat. # EACL-100; BioAssay Systems, Hayward, CA, USA).
Immunofluorescence staining and confocal microscopy
Cells were fixed with 4% paraformaldehyde and permeabilized using 0.5% Triton X-100 (Sigma) dissolved in PBS. The cells were labeled with the following primary antibodies (1:200~1:1000 dilution): anti-TrkA (cat. # 06-574; EMD Millipore), anti-ChAT (cat. # AB144P; EMD Millipore), anti-TUJ1 (cat. # MAB1637; EMD Millipore), anti–tyrosine hydroxylase (TH; cat. # 2792; Cell Signaling Technology), anti–neural cell adhesion molecule 1 (NCAM1; cat. # CBL275; EMD Millipore), anti–myelin basic protein (MBP; cat. # AB980; EMD Millipore), anti–oligodendrocyte marker O4 (cat. # O7139; Sigma), anti–neurofilament-M (NF-M; cat. # AB1987; EMD Millipore), antinestin (cat. # MAB5326; EMD Millipore), or anti–γ-aminobutyric acid (GABA; cat. # Ab86186; Abcam, Cambridge, MA, USA) overnight at 4°C. Later, the cells were incubated for 1 h with an Alexa Fluor 488– or Alexa Fluor 594–conjugated antibody (Molecular Probes, Carlsbad, CA, USA) (1:300 dilution, secondary antibodies). The cells were next stained with 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes) for 1 min. After mounting, the samples were visualized by means of a Zeiss LSM510 Meta Confocal Imaging System (Carl Zeiss, Thornwood, NY, USA).
Treatment with an ACh antagonist or agonist
α-Bungarotoxin (α-BTX; cat. # 203980; EMD Millipore), an α7 nAChR antagonist was added to the medium at a concentration of 1 μM. Cholinergic agonists ACh chloride (ACh-Cl; cat. # A6625; Sigma) and carbachol (cat. # 212385; EMD Millipore) were used at the concentrations of 1 nM and 10 pM, respectively.
Statistical analysis
Data are expressed as means of individual measurements± standard deviations. One-way analysis of variance (ANOVA) was conducted for group comparisons. Results were considered significant when p values were <0.05. Statistical analyses were performed in SPSS-19 software (IBM SPSS).
Results
Inflammatory conditions induce neuronlike morphological features in human MSCs
Human MSCs effectively inhibit lymphocyte proliferation when cocultured under MLR conditions (15, 16). In this study, human MSCs efficiently inhibited the proliferation of alloantigenically activated PBMCs (lymphocytes) acquired from various donors (Fig. 1a). Cocultured MSCs underwent extensive morphological changes such as cell thinning, remarkable elongation, filamentation, and branching (Fig. 1c) as well as enlargement of the cell body and formation of multiple neuronlike processes that appeared to mediate cell contacts with neighboring cells (Fig. 1d). Similar results were observed in another inflammatory model in which PBMC proliferation was stimulated with PHA (a mitogen). In this model, MSCs blocked lymphocyte proliferation (Fig. 1b), underwent similar neuronlike morphological changes (Fig. 1c), and grew neuronlike cell processes (Fig. 1e). To test whether the inflammation-induced neuronlike changes in MSCs were mediated by soluble factors, we performed assays in Transwell plates. MSCs and MLR-activated PBMCs were seeded in the bottom wells and insert-containing wells, respectively. After incubation for 3 days, some MSCs in the bottom wells acquired neuronlike morphological features with MLR-activated PBMCs in the insert well (Fig. 2a). These morphological changes in MSCs were not observed in the absence of activated PBMCs in the insert well (Fig. 2a). MSCs incubated with CM obtained from MLR and PHA-stimulation showed the morphological changes (Fig. 2b and Supplementary Fig. S1). MSCs incubated with the CM from activated-PBMC cultures expressed ChAT, NCAM1, NF-M, and TUJ1 (Fig. 2c).

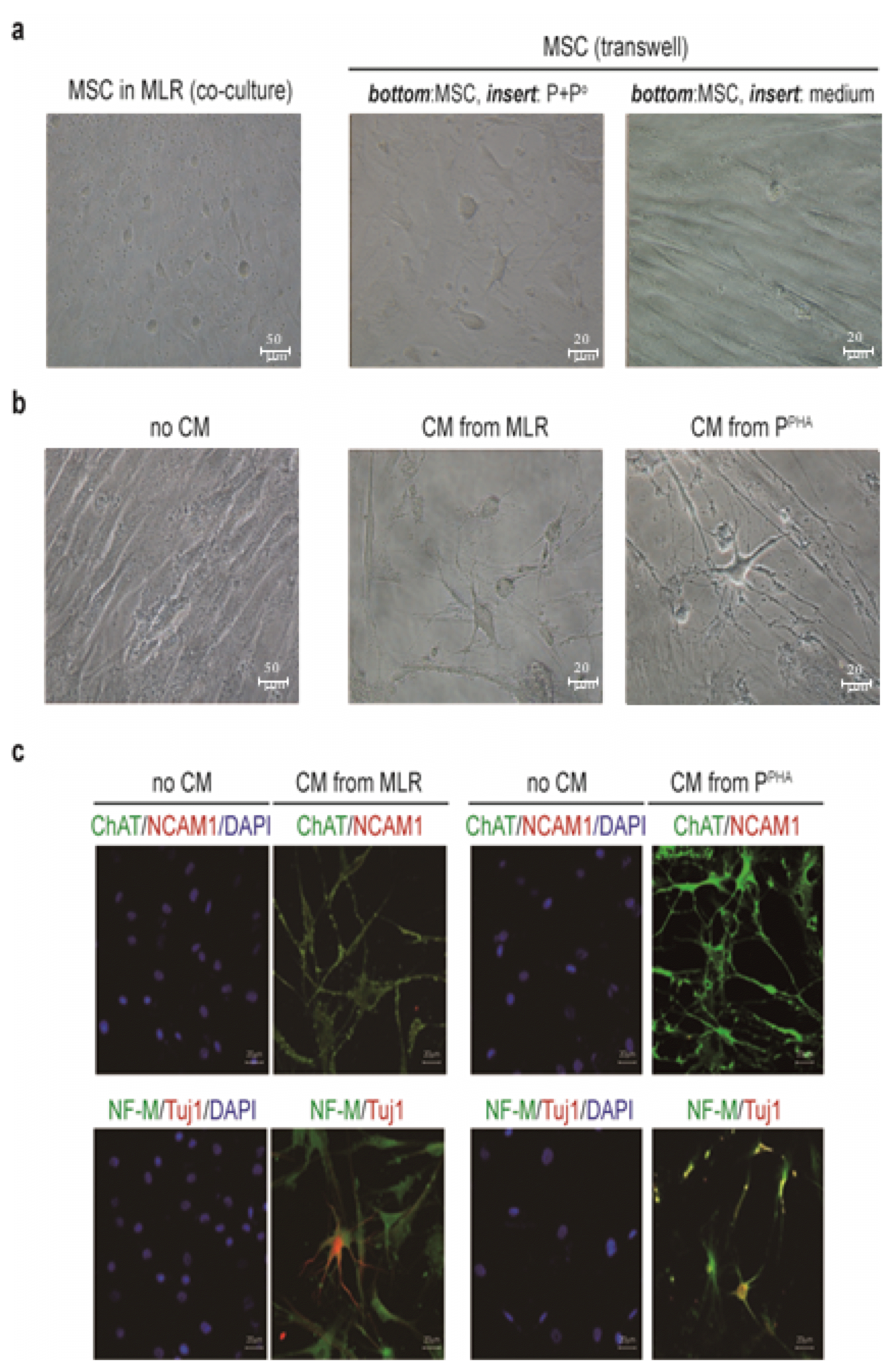
Fig. 1
Inflammatory conditions induce neuronlike morphological features in MSCs. (a) When cocultured in an MLR with PBMCs obtained from two individuals (P and Po), hBM MSCs suppressed the lymphocyte proliferation. (b) When cocultured with human PBMCs activated by PHA stimulation (PPHA), MSCs inhibited the lymphocyte proliferation. CPM: counts per million. (c) Morphological changes in MSCs were observed in cocultures with activated PBMCs. (d, e) Neuronlike morphological changes in MSCs after coculture with activated PBMCs for 48 h. (f, g) Floating neurosphere-like cell clusters (red arrows) were observed under both inflammatory conditions. Error bars are indicative of standard deviations.
![]()
Fig. 2
Inflammation induces the expression of NRs and neuron- or NPC-related markers but not glial markers on the MSC surface. (a) To determine whether inflammation-induced neuronlike phenotypic changes in MSCs were mediated by soluble factors, we performed assays using Transwell plates with inserts (0.4 μm pore size). MSCs (105) were seeded in the bottom well, and MLR-activated PBMCs (106) were seeded in the insert-containing well. After incubation for 3 days, MSCs at the bottom went through neuronlike morphological changes. These changes were not observed in the absence of activated PBMCs in the insert. (b) When MSCs were incubated for 3 days with the CM from activated PBMCs, they acquired neuronlike morphology. (c) MSCs incubated with the CM from activated PBMCs expressed ChAT, NCAM1, NF-M, and TUJ1.
![]()
Inflammation induces the expression of neurotrophin receptors (NRs) in MSCs
Furthermore, we determined whether the neuronlike morphological changes observed in MSCs were associated with neuronal marker expression. After MSCs were cocultured with activated PBMCs stimulated with MLR or PHA for 24 h, nonadherent PBMCs were removed by thorough washing, and total RNA was extracted only from adherent MSCs. The inflammatory conditions induced mRNA expression of neuronal markers TUJ1 and microtubule-associated protein 2 (MAP2) but not GFAP (Fig. 3a). Moreover, the induction of nestin, which is expressed exclusively in neuronal precursor cells (NPCs) but not in differentiated neurons (25), was evident in MSCs (Fig. 3a). Neuronal marker NF-M was not detected in MSCs (Fig. 3a). Similarly, flow-cytometric analysis confirmed the expression of proteins nestin and TUJ1 but not GFAP (Fig. 3b). Finally, immunofluorescence staining of MSCs cocultured with activated PBMCs revealed the expression of a neuronal marker (TUJ1) as well as NPC markers (nestin and NCAM1) but not a glial astrocyte marker (GFAP) or oligodendrocyte marker (O4; Fig. 3c and 3d). Taken together, these results suggested that inflammatory conditions caused MSCs to acquire neuronlike cell morphology and to express NPC markers such as nestin, TUJ1, and MAP2.
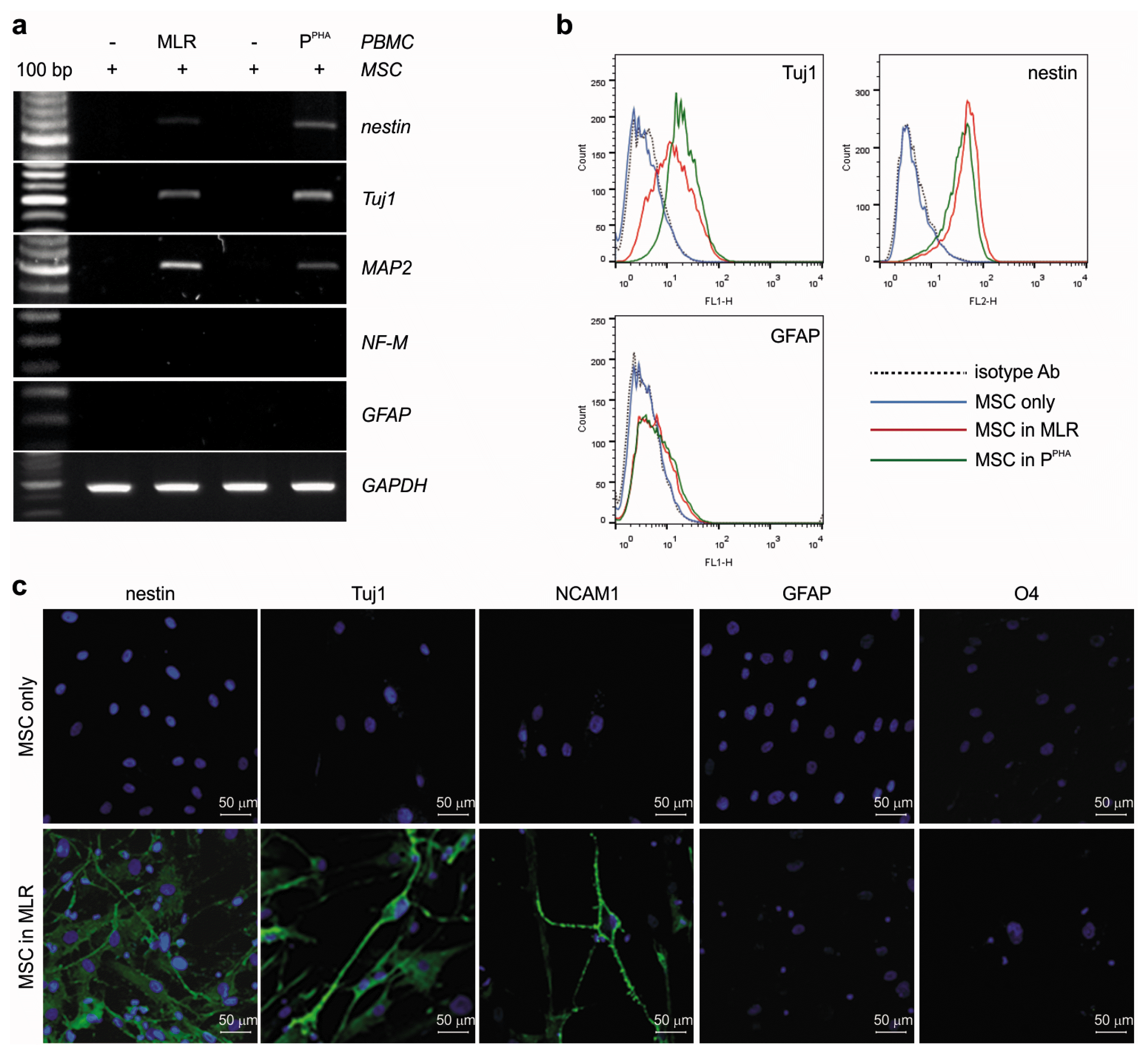
Fig. 3
Inflammation induces the expression of NRs and neuron- or NPC-related markers but not glial markers on the MSC surface. (a) RT-PCR was carried out to assess the expression of nestin, Tuj1, MAP2, NF-M, and GFAP in adherent MSCs cocultured with activated PBMCs (24 h). GAPDH served as a loading control. (b) Flow-cytometric analysis of TUJ1, nestin, and GFAP in adherent MSCs cocultured with activated PBMCs (24 h). (c) Immunofluorescence staining of nestin, TUJ1, NCAM1, GFAP, and O4 in MSCs cocultured in the MLR for 48 h. (d) Immunofluorescence staining for nestin, TUJ1, NCAM1, GFAP, and O4 in MSCs cocultured with PHA-activated PBMCs for 48 h. DAPI was used to stain nuclei. (e) RT-PCR analysis of TrkA, TrkB, TrkC, and p75NTR expression in adherent MSCs cocultured with activated PBMCs (24 h). Tuj1 served as a control for an MSC response to inflammation, and GAPDH was employed as a loading control. (f, g) qPCR was conducted to assess the expression of the aforementioned NRs as in (e). (h) Protein expression of TrkA and p75NTR, but not TrkC, was validated by western blotting in the whole MSC extracts (20 μg) after 48 h of coculture. (i, j) TrkA induction in MSCs in response to experimental inflammation was confirmed by immunofluorescence staining. Error bars are indicative of standard deviations.
![]()
![]()
We hypothesized that the acquisition of neuronlike morphology and NPC marker expression by MSCs in the inflammatory milieu was triggered at least partly by NR signaling (26). To determine whether NRs were induced in MSCs, we examined mRNA expression of the Trk gene family (A, B, and C) and p75NTR in MSCs cocultured with AICs. TrkA and p75NTR were highly upregulated, while TrkB and TrkC were not detectable after MLR or PHA stimulation (Fig. 3e and 3f). Northern and western blot analyses confirmed the expression of TrkA and p75NTR but not TrkC (Fig. 3g and 3h). Immunofluorescence staining confirmed TrkA expression under both inflammatory conditions (Fig. 3i and 3j).
Inflammation induces neurotrophins in AICs
Mitogen-activated human T lymphocytes have been reported to secrete remarkable quantities of NGF, a well-characterized neurotrophin (27). Accordingly, we determined whether inflammation could upregulate neurotrophins in AICs. The mRNA expression of NGF and BDNF was highly upregulated in immune cells during MLR (Fig. 4a and 4b) or under PHA stimulation conditions (Fig. 4c and 4d). The secretion of NGF and BDNF from immune cells under MLR (Fig. 4e and 4f) or PHA stimulation condition (Fig. 4g and 4h) was confirmed by ELISA. Therefore, inflammatory conditions induced expression of NRs on the surface of MSCs as well as neurotrophins in AICs.
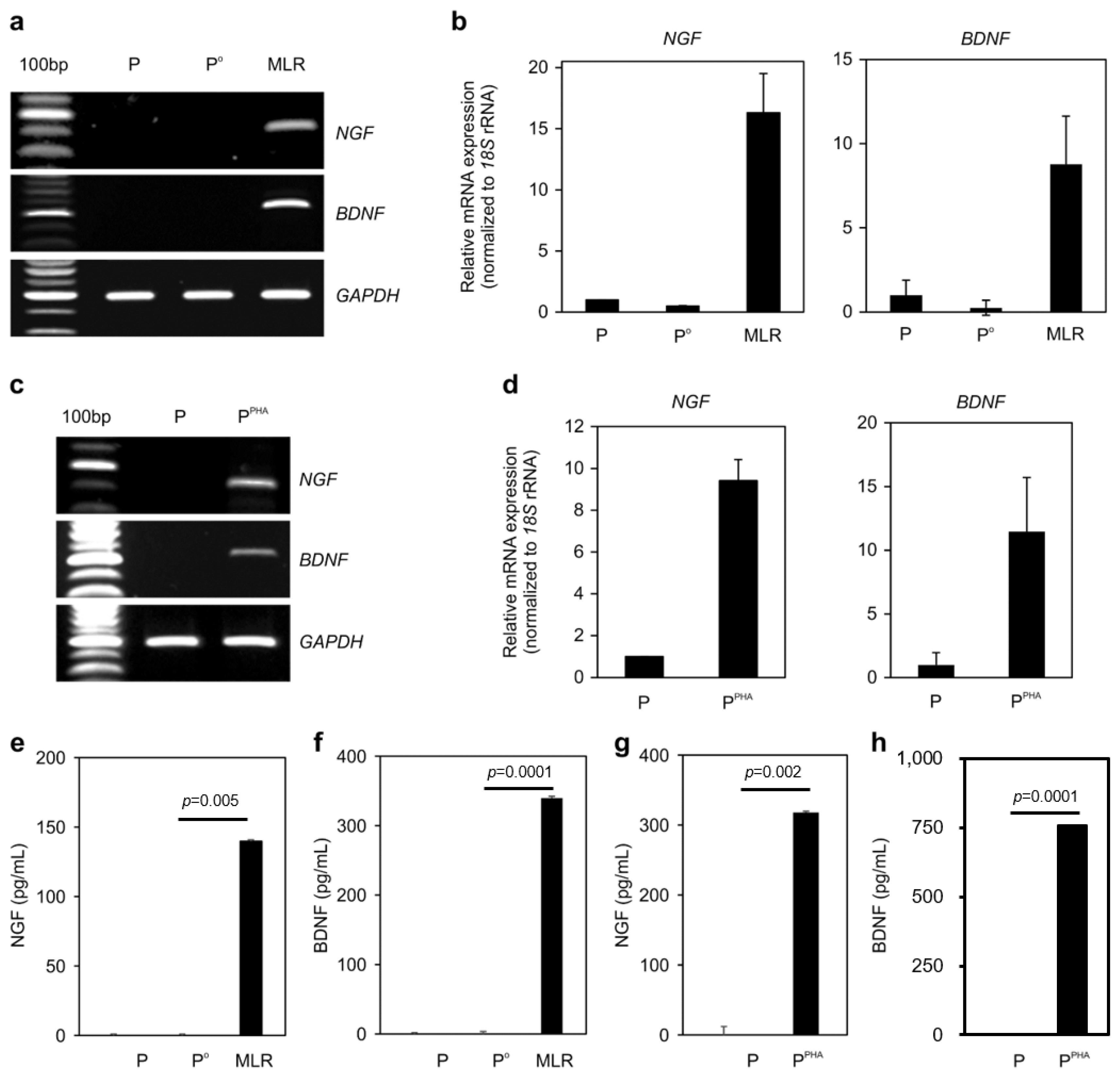
Fig. 4
Neurotrophins are induced in PBMCs activated by MLR or PHA. (a) RT-PCR analysis of NGF and BDNF expression in PBMCs after activation by MLR for 24 h. (b) qPCR analysis of NGF and BDNF expression in the samples as described in panel (a). (c) RT-PCR analysis of NGF and BDNF expression in PBMCs after stimulation with PHA for 24 h. (d) qPCR analysis of NGF and BDNF expression in the samples as described in panel (c). (e~h) ELISA quantification of soluble NGF and BDNF in the CM obtained from activated-PBMC cultures (48 h). Three independent experiments were conducted.
![]()
MSCs with inflammation-induced neuronlike features release ACh and mediate immunosuppression via the ACh–nAChR signaling pathway
Next, to characterize the acquired properties and to understand the neuronlike morphological changes in MSCs, we tested whether these cells produced and released neurotransmitters. Under inflammatory conditions, MSCs developed neuronlike morphology and expressed ChAT (but not GABA) or TH (Fig. 5a). The ChAT expression was confirmed at mRNA and protein levels (Fig. 5b and 5c). The aforementioned induction probably involves soluble factors owing to the neurotrophin upregulation in AICs. Upon treatment of MSCs with the CM from PHA stimulation condition (PPHA) or MLR for 48 h, we detected approximately 4- to 5-fold greater ACh secretion by MSCs compared to that in AICs in the absence of MSCs (Fig. 5d). When new MSCs were treated with the CM from MLR supernatant with old MSCs, additional ACh secretion was detected (Fig. 5e), suggesting that new MSCs were able to secret additional ACh upon treating with CM from MLR with old MSCs.
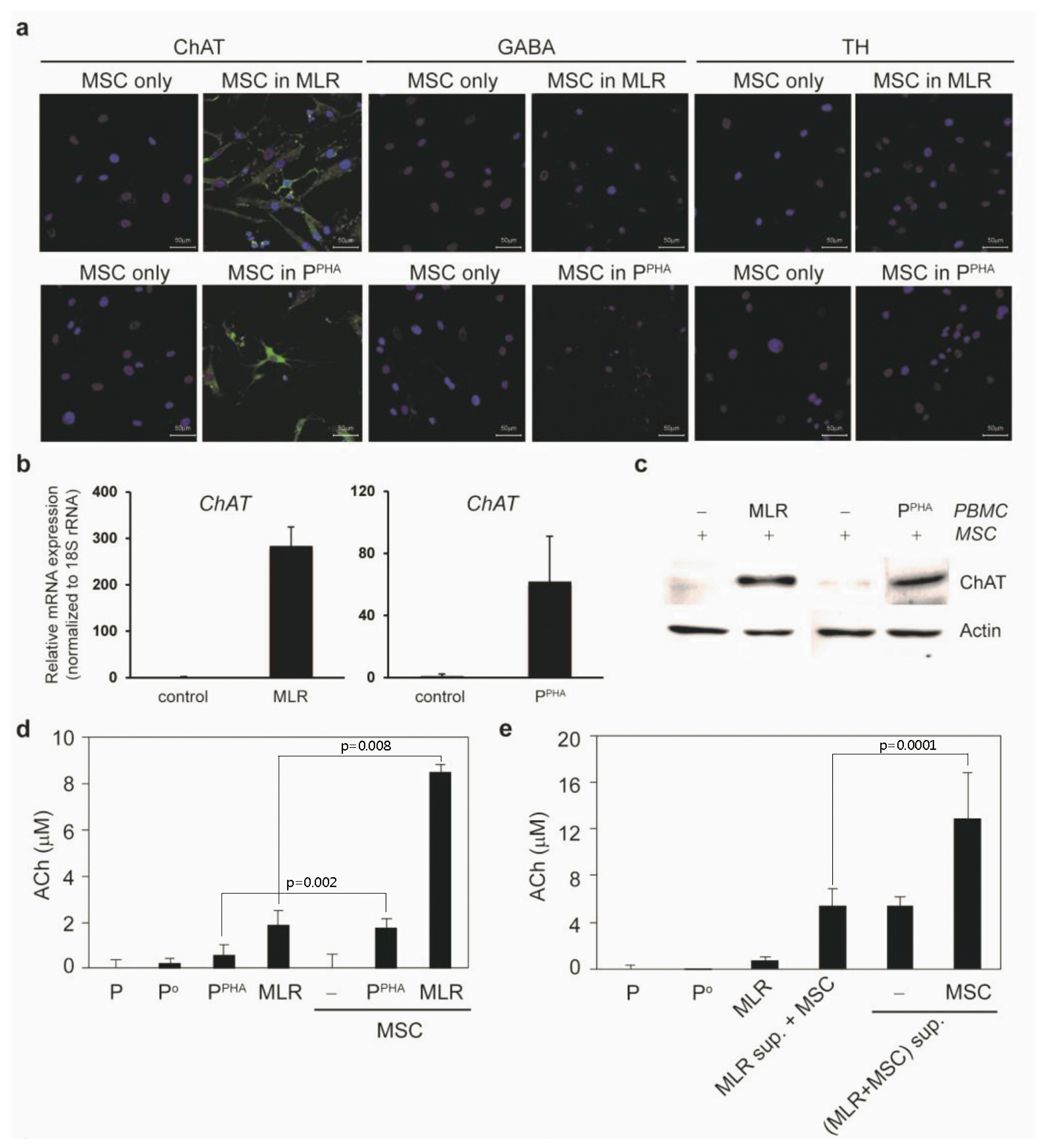
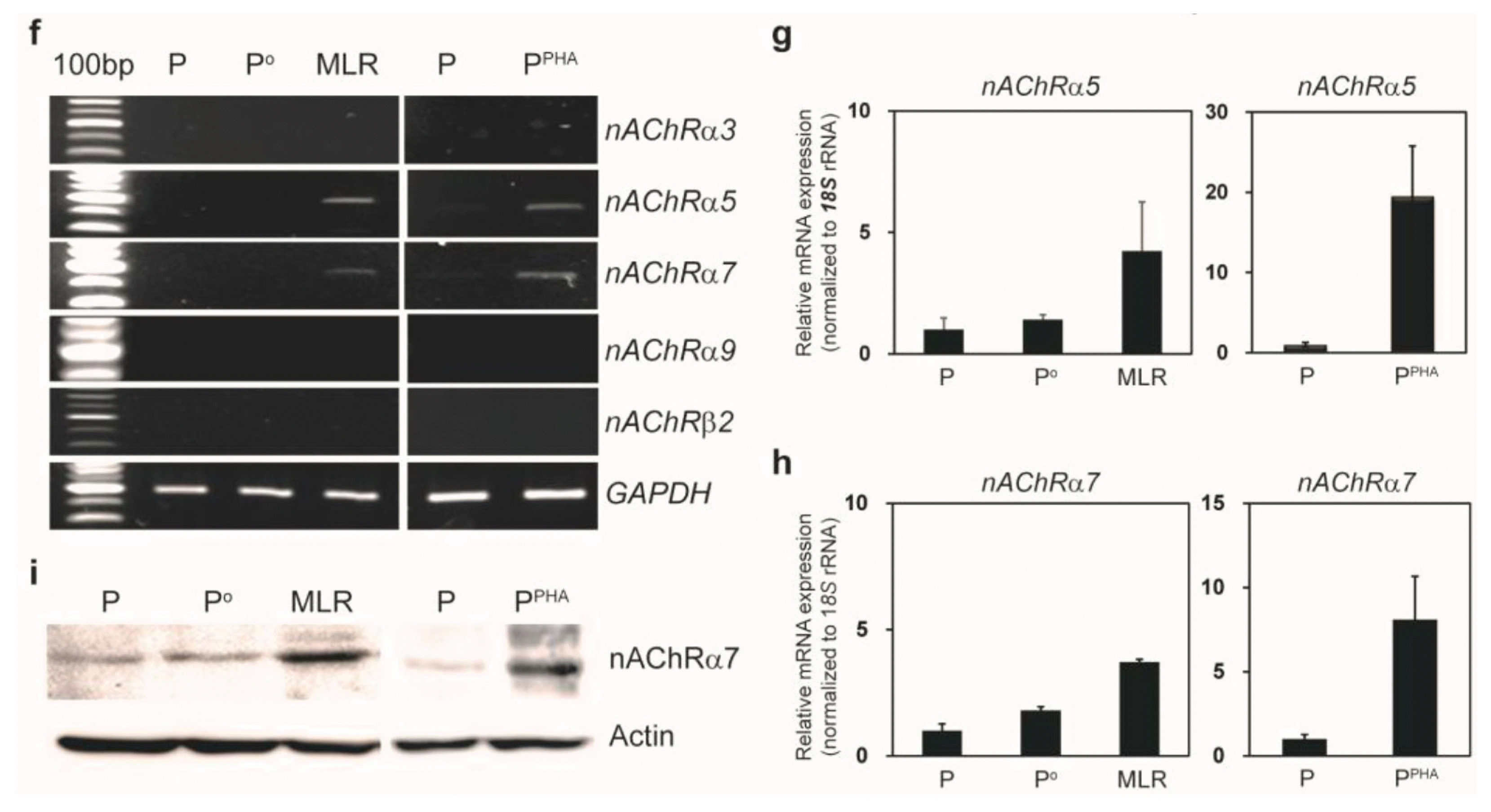
Fig. 5
Inflammatory conditions induce a cholinergic-neuron–like phenotype in MSCs and nAChRs in activated PBMCs. (a) Immunofluorescence staining of ChAT, GABA, and TH in MSCs cocultured with activated PBMCs (48 h). (b) qPCR was carried out to quantify ChAT expression in MSCs after inflammatory stimulation for 24 h. (c) Western blotting confirmed ChAT expression in the whole MSC extract (20 μg) after coculture for 48 h. (d) ACh and choline concentration was measured in the CM obtained from PBMCs alone (P or Po), PHA-activated PBMCs (PPHA), or MLR (P and Po) culture without or with MSCs (n=3). (e) ACh and choline concentration was measured in the CM obtained from PBMCs alone (P or Po) or MLR (P and Po) culture without or with MSCs for 48 h. “MLR sup.” is the supernatant from the MLR without MSCs. “(MLR+MSC) sup.” is the supernatant from MLR with MSCs (n=3). (f) RT-PCR analysis of several nAChR subunits in activated PBMCs. (g) qPCR was carried out to assess nAChR α5 expression in activated PBMCs after incubation for 24 h. (h) qPCR was performed to measure nAChR α7 expression in activated PBMCs after incubation for 24 h. (i) An increase in the protein expression of the nAChR α7 subunit in activated PBMCs was confirmed by western blotting of whole MSC extracts prepared after MLR or PHA stimulation for 48 h.
![]()
![]()
We determined whether AICs expressed ACh receptors that allowed for binding of ACh (released by neuronlike MSCs) and thus for inhibition of AICs. Screening to detect muscarinic and nicotinic receptor family members revealed that inflammatory stimulation induced mRNA expression of nAChR subunits α5 and α7 (Fig. 5f~h) as well as protein expression of the nAChR α7 subunit (Fig. 5i) in PBMCs under MLR and PHA stimulation conditions. Corresponding mRNA expression of muscarinic receptors was not observed (data not shown). Little or no nAChR α7 was detected on the surface of MSCs under inflammatory conditions (Supplementary Fig. S2), suggesting that ACh released by MSCs did not exert an autocrine effect on MSCs.
MSC-mediated immunosuppression via ACh is reversed by α-BTX (an ACh antagonist), whereas lymphocyte proliferation is inhibited by ACh-Cl (an ACh agonist)
To demonstrate the involvement of an inhibitory mechanism in neuronlike-MSC–mediated immunosuppression, α-BTX (an antagonist of nAChR α7) was added to the medium. Incubation with α-BTX effectively restored the MSC-blocked lymphocyte proliferation (Fig. 6a and 6d) and rescued the MSC-suppressed TNF-α and IFN-γ secretion after MLR (Fig. 6b and 6c) or PHA stimulation (Fig. 6e and 6f). After that, we examined whether AChR agonists could mimic the immunosuppressive function of MSCs by inhibiting immune-cell activation. Treatment with an AChR agonist (i.e., ACh-Cl) significantly inhibited the proliferative response of MLR- or PHA-stimulated PBMCs (Fig. 6g and 6j) and suppressed the TNF-α and IFN-γ secretion by AICs (Fig. 6h, 6i, 6k, and 6l). Similar results were obtained with the nonspecific cholinergic agonist carbachol (Supplementary Fig. S3). Collectively, these data meant that neuronlike MSCs employed the ACh–nAChR signaling pathway to modulate the inflammatory response of immune cells.
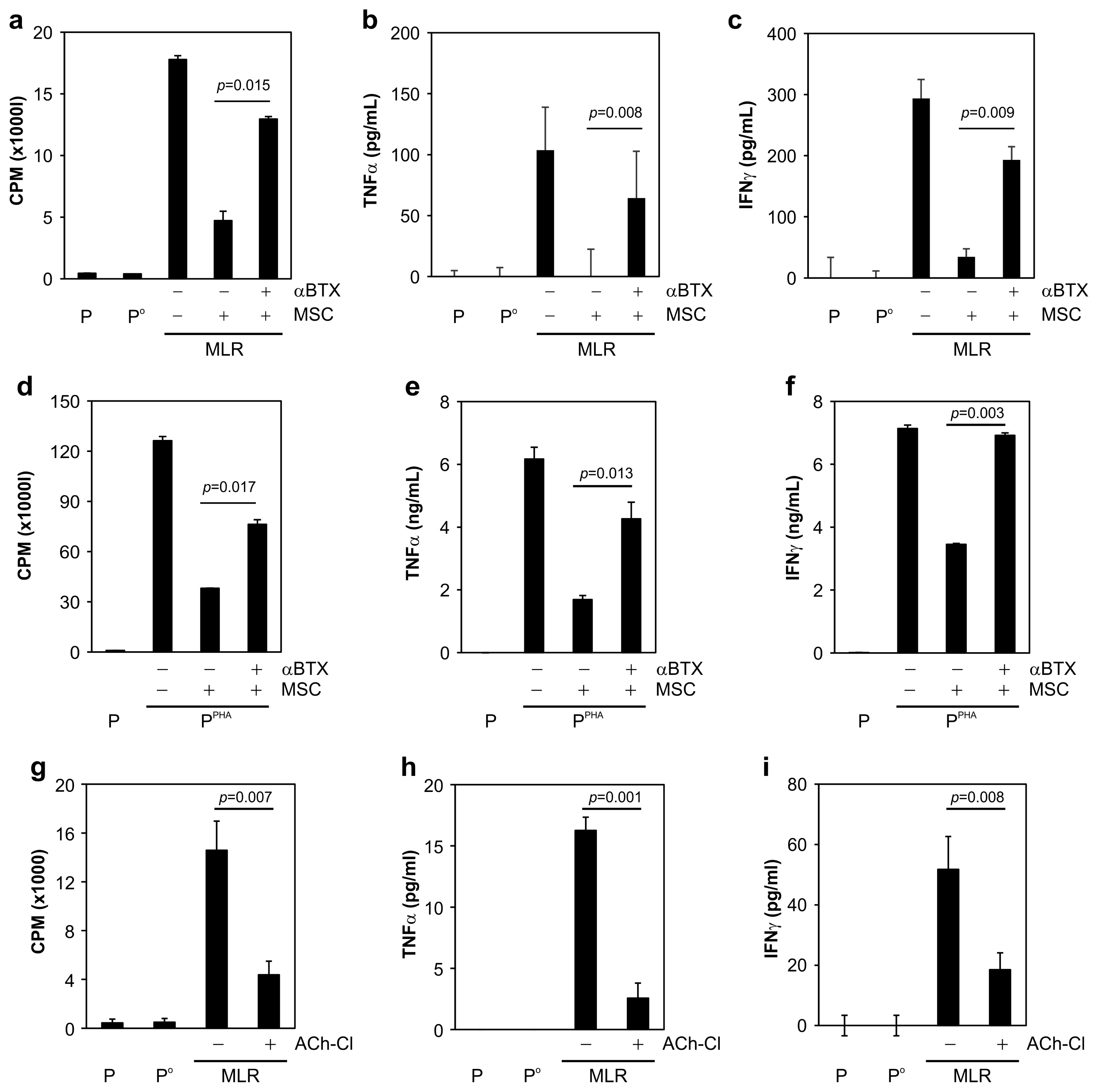
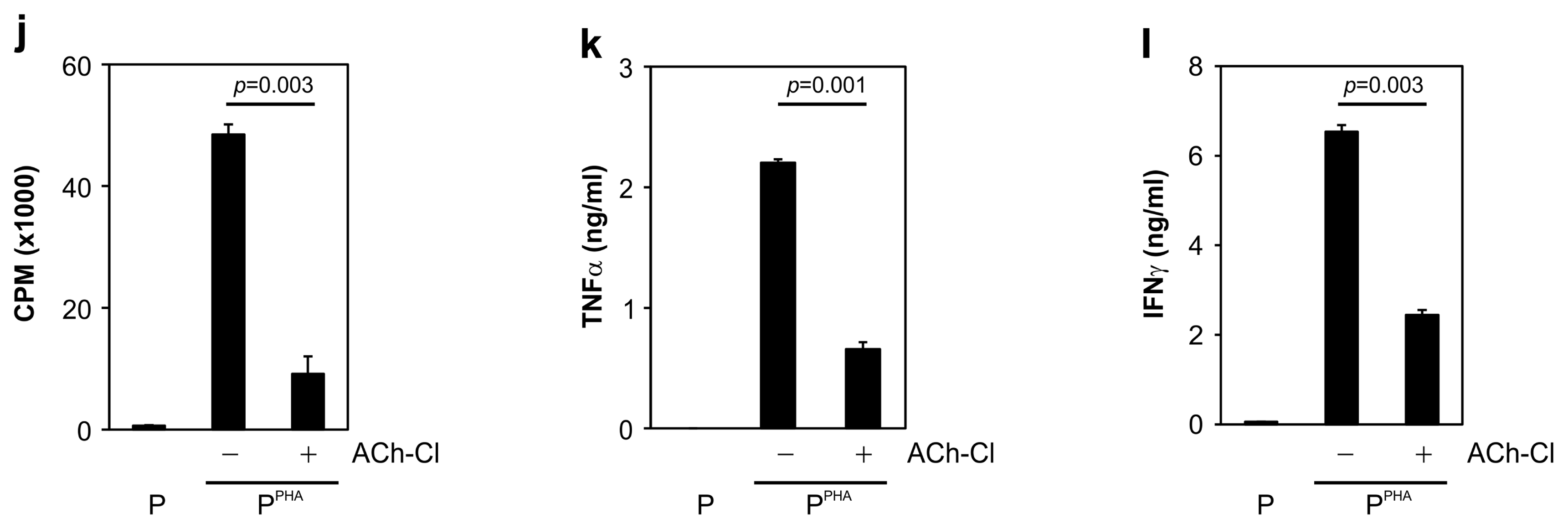
Fig. 6
MSC-mediated immunosuppression via ACh is reversed by α-BTX (ACh antagonist), and lymphocyte proliferation is inhibited by ACh-Cl (ACh agonist). (a) MSC-suppressed lymphocyte proliferation was significantly restored by α-BTX addition to the MLR medium. (b) MSC-suppressed TNF-α production during MLR was restored by α-BTX treatment. (c) Suppressed secretion of IFN-γ during MLR was significantly restored by α-BTX treatment. (d) MSC-mediated suppression of PHA-stimulated lymphocyte proliferation was significantly attenuated by α-BTX treatment. (e) MSC-suppressed TNF-α production in PHA-activated PBMCs increased in the presence of α-BTX. (f) The reduced IFN-γ secretion by PHA-activated PBMCs was restored by α-BTX. (g, j) ACh-Cl addition to the medium significantly attenuated the increase in lymphocyte proliferation caused by (g) MLR or (j) PHA treatment. (h, k) Production of TNF-α by activated PBMCs elicited by (h) MLR or (k) PHA stimulation was attenuated by ACh-Cl treatment. (i, l) ACh-Cl treatment significantly suppressed the IFN-γ secretion from PBMCs activated by (i) MLR or (l) PHA stimulation. All data are the average of 3~5 independent experiments and were statistically evaluated by paired Student’s t test. p values <0.05 were assumed to indicate statistically significant variations.
![]()
![]()
Discussion
The immunosuppressive function of MSCs distinguishes them from the other types of stem cells such as embryonic stem cells and induced pluripotent stem cells. This property is important because a successful clinical application of stem cells requires overcoming an immune response in the host. MSCs were studied here owing to their ability to attenuate an exacerbated inflammatory response and autoimmunity (7). Although several studies have shown therapeutic effects of MSCs on autoimmune disorders such as graft-versus-host disease, Crohn’s disease, and multiple sclerosis (8, 12), the precise mode of action of MSCs as immunomodulators is not well understood. Nevertheless, there exists a prevalent notion that ex vivo–cultured MSCs have an immunosuppressive function and are beneficial to patients with various autoimmune diseases.
In this study, we provide evidence for a new immunosuppressive mechanism employed by MSCs. We demonstrate that the acquired neuronlike morphology of MSCs during inflammation may be related to inhibition of lymphocyte proliferation via ACh secretion. Our observations indicate that the immunosuppressive activity of MSCs depends on cell–cell signaling via direct contact as well as soluble factors such as indoleamine 2,3-dioxygenase, HLA-G5, prostaglandin E2, TSG-6, and NO (6, 12, 13). Given that the anti-inflammatory functions of these factors remain undefined in specific pathologies, unknown players may be involved in MSC-mediated immunosuppression. It is necessary to investigate whether any other immunomodulatory factors interact with ACh or play a compensatory role in MSC-mediated immunosuppression.
Our findings support other reports about cholinergic receptors on immune cells, whose activation suppresses inflammatory responses (18, 21, 28). Human immune cells including leukocytes and lymphocytes express ChAT, AChR, and AChE (28). ACh in blood is released mainly by T lymphocytes because ChAT is not expressed in B lymphocytes (18). ACh is a primary parasympathetic neurotransmitter and is synthesized by preganglionic fibers of the sympathetic nervous system and parasympathetic nervous system (PSNS) and by postganglionic PSNS fibers (29). In this study, to the best of our knowledge, we demonstrate for the first time ACh secretion by MSCs under inflammatory conditions. Only a low concentration of ACh was secreted by immune cells during MLR. In contrast, approximately a 4- to 5-fold larger amount of ACh was secreted by MSCs treated with the CM from MLR or PHA-stimulated culture in comparison with AICs (Fig. 5d~e). This result strongly indicates that neuronlike MSCs secrete ACh, which results in the suppression of immune cells during inflammatory conditions.
Recent studies indicate that EVN signaling (a major component of the PSNS of the ANS) inhibits the production of proinflammatory cytokines and systemic inflammation (30). This regulation requires an interplay between EVN signaling and nAChR expressed on immune cells (20). Many reports have proved the importance of nAChR α7 in cholinergic anti-inflammatory signaling (21), implying strong dependence of this mechanism on this receptor. nAChR α7 is expressed by a variety of non-neuronal cell types including macrophages, endothelial cells, and lymphocytes (22, 29, 31, 32). Various studies have proved the functional importance of nAChR α7 expression on the surface of CD4+ T lymphocytes (19, 28, 33). Consistent with the aforementioned findings, our results indicate that the MLR- or PHA-dependent activation of immune cells stimulates the expression of nAChR subunits α5 and α7 (Fig. 5f~i), potentiating the cholinergic feedback pathway in immune cells. MSC-mediated suppression of the proliferation and function of immune cells was significantly inhibited by the nAChR α7 antagonist (α-BTX). These data mean that MSC-mediated immunosuppression is driven by cholinergic inhibitory signaling. α-BTX binds to neuronal nAChR subunits such as α7 and α9 as well as muscle-type nAChR (33). Given that T lymphocytes express neither muscle-type nAChR nor nAChR α9, α-BTX–mediated inhibition probably predicts the involvement of nAChR α7–regulated cholinergic signaling in MSC-mediated immunosuppression (19, 33, 34). Because one report indicates that nAChR α7 is detectable in MSCs, we examined the expression of nAChR α7 on the MSC surface under inflammatory conditions. The induction of nAChR α7 was weak or absent in MSCs (Supplementary Fig. S2), suggesting that the autocrine effect on MSCs is probably insignificant.
In this study, the CM of AICs induced a neuronlike phenotype in MSCs. Therefore, the soluble factors responsible for this phenotypic change are probably released by AICs. Although neurotrophins are expressed and released primarily by the nervous system, a few of them, such as NGF and BDNF, are produced by AICs, including human CD4+ T lymphocytes (27, 35, 36). In accordance with those other findings, our results indicate that inflammatory conditions elicit immediate expression of NGF and BDNF by AICs. The simultaneous expression of neurotrophins by AICs and upregulation of NRs in MSCs provide mechanistic clues to the inflammation-induced neuronlike phenotype of MSCs. In contrast, the treatment of MSCs with recombinant NGF and BDNF in combination or alone did not induce ACh secretion by MSCs in vitro (Supplementary Fig. S4), implying that some factors other than NGF and BDNF must be involved in the additional secretion of ACh by MSCs.
Our findings suggest that immunosuppressive activity might be mediated by ACh secreted from neuronlike MSCs in the inflammatory milieu. An inflammatory environment in which neurotropic factors such as NGF and BDNF are secreted by AICs causes MSCs to assume a neuronlike phenotype. This change in turn leads to the ACh release from MSCs in the presence of NGF, BDNF, and unknown stimulatory factors and inhibits lymphocyte proliferation and proinflammatory-cytokine production in AICs through the ACh–nAChR signaling pathway (Fig. 7). We can speculate that these events may take place in secondary lymphoid organs such as lymph nodes. Previously, we have reported that MSCs migrate to the lymph nodes near inflamed tissues (11). We can hypothesize that MSCs migrate to the draining lymph nodes where AICs are produced to set up a proinflammatory milieu suitable for the induction of neuronlike features in MSCs followed by the inhibition of lymphocyte proliferation and activities through the ACh–nAChR interaction. Considering that a small number of injected MSCs is sufficient to reduce systemic inflammation, it is possible that MSCs impose efficient immunosuppression within the draining lymph nodes in which a few MSCs can regulate a relatively large number of AICs via the ACh–nAChR interaction. Other unknown released factors in addition to ACh from neuronlike MSCs together may play important roles in the regulation of inflammation and amelioration of immune diseases.
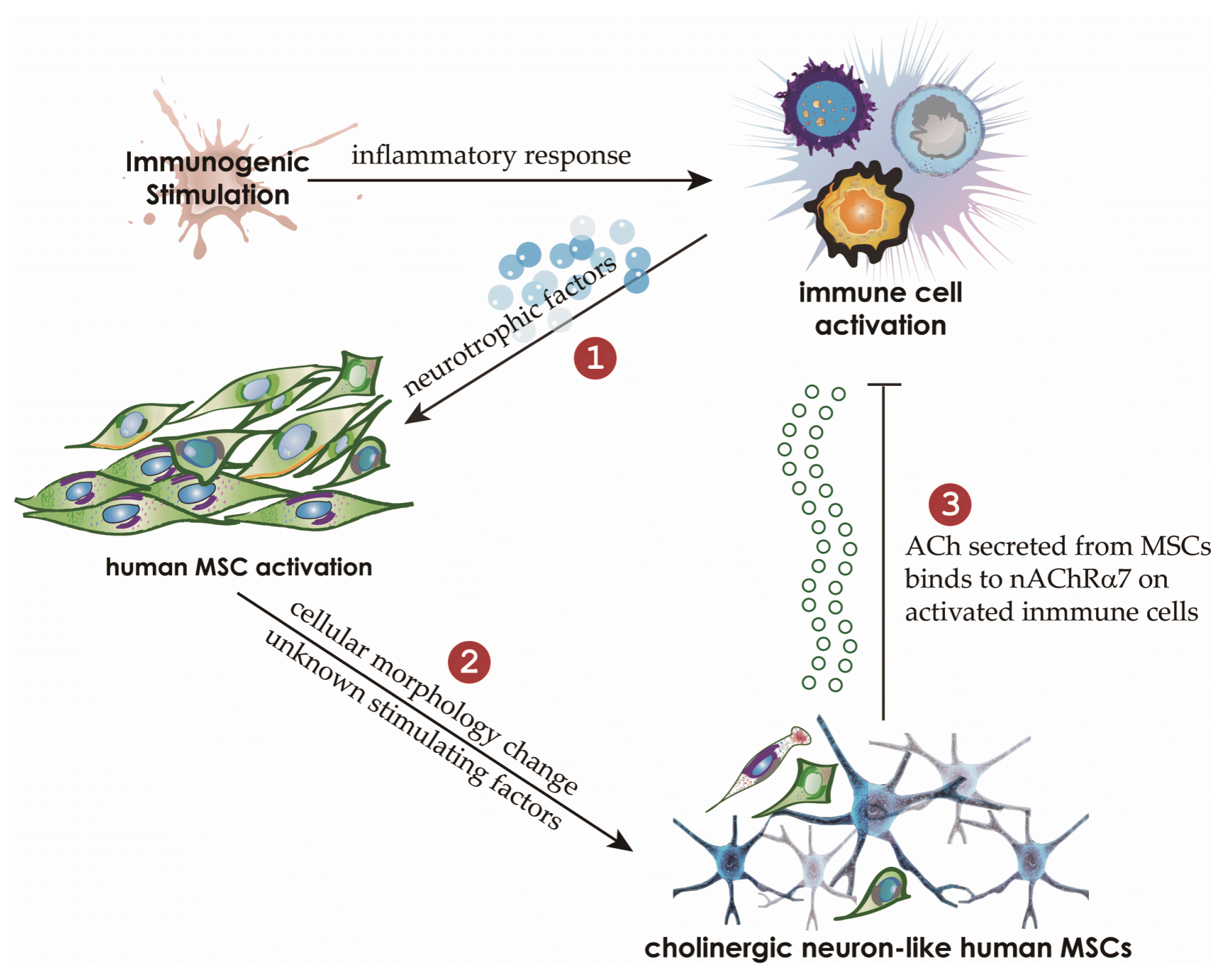
Fig. 7
The proposed model of an immunomodulatory mechanism utilized by human MSCs in an inflammatory milieu. The inflammatory conditions drive human MSCs to adopt a neuronlike phenotype. It is probable that the expression of neurotrophins such as NGF and BDNF in human AICs and the presence of NRs on MSCs are associated with these changes in MSCs. Furthermore, the inflammatory milieu probably induces the expression of nAChR including nAChR α7, which participates in the negative regulation of activated lymphocytes. Neuronlike MSCs stimulated by neurotrophins and unknown factors secrete ACh, which binds to AICs via nAChR α7, thereby inhibiting the proliferation and function of AICs.
![]()
Conclusions
Our findings point to a novel mechanism of human-MSC–mediated immunosuppression. The inflammatory milieu created by immune-cell activation causes MSCs to manifest neuronlike characteristics and to release ACh, which then inhibits inflammatory responses via the ACh–nAChR signaling pathway. A detailed understanding of the biological mechanism underlying MSC-mediated immunosuppression is expected to lead to the development of therapeutics involving MSCs for autoimmune and/or inflammatory disorders.
 XML Download
XML Download