

 PDF
PDF Citation
Citation Print
Print


Introduction
The repair capacity of articular cartilage after injury or disease is poor. Mesenchymal stem cells are promising candidates for the use of cell based tissue engineering applications for the repair of cartilage tissue lesions. The chondrogenic potential of MSCs has been shown in different matrix free and matrix based cell culture systems (1–7). In the commonly used pellet culture system, MSCs differentiate chondrogenically in a serum free medium containing TGFβ and dexamethasone (6). However, chondrogenic differentiating MSCs express hypertrophy markers like collagen type X, alkaline phosphatase (ALP) and MMP-13 (8–11). This behaviour of chondrogenic differentiating MSCs resembles that of growth plate chondrocytes during endochondral ossification. During endochondral bone development growth plate chondrocytes do not rest on a developmental stage typical for articular chondrocytes but spontaneously proceed towards the hypertrophic stage. Hypertrophic chondrocytes then undergo apoptosis, the tissue is invaded by blood vessels and osteoprogenitor cells and bone is formed. Vascular invasion and matrix calcification has also been observed after in vivo transplantation of human chondrogenic MSC pellet cultures into mice (12, 13). This biological behaviour of chondrogenic differentiating MSCs raises concern for a tissue engineering application of MSCs in articular cartilage repair. It is important to better understand the mechanisms that regulate late differentiation steps in chondrogenic differentiating MSCs to find ways to inhibit hypertrophy. The similarity of MSC chondrogenesis and embryonic endochondral ossification indicates that similar mechanisms are involved in both biological processes (10).
TGFβ signaling has been shown to play a crucial role in the regulation of endochondral ossification. In vivo and in vitro studies showed that TGFβ signaling promotes chondrogenic differentiation of mesenchymal cells and embryonic chondrocytes (14–18). In addition, TGFβ signaling is important in the regulation of chondrocyte maturation. TGFβ signaling inhibits hypertrophy in vitro and in vivo. In vitro studies showed that TGFβ inhibits hypertrophy and the expression of hypertrophic markers like collagen type X and ALP in cultured embryonic chondrocytes (19–21). In vivo it was shown that the application of TGFβ into a developing chick limb inhibits chondrocyte hypertrophy and loss of function models of TGF β signaling result in premature chondrocyte hypertrophy in mice (22–24).
TGFβ1, 2 and 3 are the classical inducers of MSC chondrogenesis (1, 7). However, the exact role of TGFβ signaling in the regulation of MSC hypertrophy is relatively unknown. Here we used an in vitro hypertrophy model for chondrogenic differentiating MSCs in which the hypertrophic phenotype can be strongly enhanced by modulations in the medium conditions. Differential expression analysis of TGFβ signaling associated genes was carried out between standard chondrogenic and hypertrophy enhancing conditions, TGFβ signaling activity was measured comparatively between the two conditions and functional experiments using TGFβ signaling modulators were conducted.
Go to : 
Materials and Methods
Isolation of MSCs
MSCs were isolated from iliac crest bone marrow aspirates of seven male patients, aged 21 to 42 years, undergoing surgery that required autologous bone grafting with approval of the local ethics committee and informed written consent. MSCs were isolated by Ficoll (Biochrom) gradient centrifugation followed by plastic adhesion. Cells were expanded in Dulbecco’s modified Eagle’s medium (DMEM) low glucose (Invitrogen) with 10% fetal calf serum (PAN Biotech GmbH) and 1% penicillin/streptomycin (Invitrogen) at 37°C with 5% CO2. The medium was changed twice a week and cells were trypsinized at 80% confluence and frozen for later use in liquid nitrogen. After thawing and monolayer expansion, cells were used for the experiments at passage 1.
Chondrogenic differentiation and enhancement of hypertrophy
MSCs were trypsinized and seeded in V-bottomed 96-well polypropylene plates at 200,000 cells per well. Pellets were formed by centrifugation at 250 g for 5 min and chondrogenically differentiated in DMEM with high glucose (Invitrogen), 1% ITS (Sigma Aldrich), 50 μg/ml ascorbate-2-phosphate (Sigma Aldrich), 40 μg/ml L-proline (Sigma Aldrich), 100 nM dexamethasone (Sigma Aldrich), 1 mM sodium pyruvat (Invitrogen) and 10 ng/ml TGFβ1 (R&D Systems).
After a pre-differentiation period of 14 days, medium conditions were changed to hypertrophy enhancing medium (hyp) consisting of DMEM high glucose, 1% ITS, 50 g/ml ascorbate-2-phosphate, 40 g/ml L-proline, 1 nM triiodothyronine (T3) (Sigma Aldrich) and the control was kept in chondrogenic medium (chon) for the whole culture period. The medium was changed three times per week.
Aggregates were harvested at d1, d3, d7, d14, d17, d21 and d28 for gene expression analysis. Aggregates for histological analysis were harvested on d14 and d28.
Modulation of hypertrophy by TGFβ-type-1-receptor inhibitor SB431542
Aggregates were differentiated chondrogenically for 14 days. On day 14, aggregates were either kept in chondrogenic medium with 0.5 μM, 3 μM or 10 μM SB431542 (Tocris) or transferred to hypertrophic medium with 0.5μM, 3 μM or 10 μM SB431542 for additional 14 days. Aggregates that were kept in chondrogenic and hypertrophic medium without SB431542 were used as a control.
In another experiment aggregates were pre-differentiated in chondrogenic medium for 14 days followed by incubation in chondrogenic medium including 100 ng/ml BMP4 (R&D systems) and 0.5 μM, 3 μM or 10 μM SB-431542.
Aggregates were histologically investigated on d28.
Histology, histochemistry and immunohistochemistry
Aggregates were harvested on d14 and d28, fixed in 4% paraformaldehyde and 10μm thick frozen sections were prepared.
Sections were stained with dimethylmethylene blue (DMMB) (Sigma Aldrich) for glycosaminoglycans. Histochemical ALP staining was performed with an alkaline phosphatase kit (Sigma Aldrich) with neutral red as counterstain.
For immunohistochemistry mouse anti collagen type X (1:20, Quartett Immunodiagnostika und Biotechnologie GmbH) and mouse anti collagen type II (1:100, Calbiochem) antibodies were used and immunohistochemistry was carried out as follows: . After blocking of endogenous peptidases (3% H2O2/ 10% Methanol in PBS) for 30 minutes, antigen retrieval with pepsin digestion for 15 minutes at room temperature (RT) was performed. Then sections were incubated in blocking buffer (10% fetal bovine serum/10% goat serum in PBS) for 60 minutes at RT followed by incubation in an appropriate primary antibody in blocking buffer overnight at 4°C. For collagen type X staining additional hyaluronidase digestion for 60 minutes at RT was performed prior to blocking. Immunolabeling was detected with a biotinylated secondary antibody (1:100; Dianova), horseradish peroxidase conjugated streptavidin (Vector Laboratories, Burlingame) and metal enhanced diaminobenzidine as substrate (Sigma Aldrich).
RNA isolation, cDNA synthesis and gene expression analysis
For each of the 7 different independent donors 8 to 10 aggregates per condition and time point were pooled for the experiments, homogenized in 1 ml TRI Reagent (Sigmal Aldrich) using the Power Gen 1000 homogenizer (Fisher Scientific) and RNA was isolated by the Trizol method. Reverse transcription was performed with Transcriptor First Strand cDNA Synthesis kit (Roche). Semi-quantitative real-time PCR was performed with Brilliant SYBR Green QPCR mix (Stratagene) and the Mx3000P QPCR System (Stratagene). Gene expression was normalized to hypoxanthine guanine phosphoribosyltransferase (HPRT).
For primers use din real time PCR please see Table 1.
Table 1
Primer sequences for real time PCR
![]()
Western blot analysis
For Western Blot Analysis, the following antibodies were used: mouse anti Sox9 (1:500, Millipore); rabbit anti TGFβR1 (1:500, Abcam); rabbit anti β actin (1:10000, Abcam); rabbit anti Smad2 (1:1000, Cell Signaling); rabbit anti Smad3 (1:1000, Cell Signaling); rabbit anti phospho-Smad2 (1:1000, Cell Signaling); rabbit anti phospho-Smad3 (1:1000, Cell Signaling).
5 to 8 MSC pellets per time point and per condition for each patient were pooled, washed in ice cold PBS and homogenized in 500 μl 6 M urea/2% SDS solution containing protease inhibitor cocktail (Sigma) and phosphatase inhibitor (Sigma) using the Power Gen 1000 homogenizator (Fischer Scientific). The lysate was centrifuged for 5 minutes at 1000 g (4°C) and the supernatant was transferred to a fresh tube. The protein concentration of the supernatant was determined using the BCA Protein Assay kit (Biorad, DC Protein Assay) according to the manufacturer’s instructions.
Lysates were supplemented with 4× LDS sample buffer (Invitrogen) and 10 mM dithiothreitol (DTT) and proteins were denatured for 5 minutes at 95°C. For gel electrophoresis, equal amounts of protein (10 μg) were loaded and separated on a 4~12% bis tris gel (Novex by Life Technologies) at 120 V. After gel electrophoresis proteins were transferred from the gel to a polyvinylidenfluoride (PVDF) membrane (Millipore). Blotting was performed for 2 hours at 100 V. After transfer, the membrane was blocked for 1 hour in 5% skim milk powder in TRIS buffered saline with Tween 20 (TBST). The membrane was then incubated in primary antibody in 5% skim milk powder in TBST over night at 4°C. The next day, the membrane was washed three times for 10 minutes in TBST and afterwards incubated in HPR-coupled secondary antibody (1:1000, Pierce) in 5% skim milk powder in TBST at room temperature for 1 hour. Then the membrane was washed three times in TBST for 10 minutes. Chemoluminescence was detected with the ECL western kit (Pierce) and by using x-ray sensitive films (ECL Hyperfilm, Amersham). The films were developed in a photo developer (Curix 60,) AGFA).
Western Blot membranes were stripped using Re-blot Plus (Millipore) according to manufactures instructions.
Statistical analysis
Gene expression was analyzed by calculating the means of every relative expression normalized to the housekeeping gene HPRT. After checking for normal distribution by Kolmogorow-Smirnoff testing a two tailed student’s t-test was used. To maintain an overall p-level of p<0.05, posthoc Bonferronis testing was carried out.
Go to :
Results
Enhancement of hypertrophy
In our study the enhancement of hypertrophy by addition of T3 was shown by an increased cell size, stronger collagen type X staining and higher ALP activity under pro-hypertrophic conditions compared to standard chondrogenic conditions. Extracellular matrix analysis revealed poorer glycosaminoglycan content (by DMMB staining, Fig. 1A, B), and immunohistochemically equal to less collagen type 2 staining (Fig. 1C, D) after hypertrophic conversion. Analysis for hypertrophic markers showed increased collagen type 10 staining (Fig. 1E, F) as well as strongly increased staining for the pre-apoptotic marker alkaline phospathase (Fig. 1G, H) for samples after hypertrophic conversion compared to non-converted samples.
 | Fig. 1Differences in glycosaminoglycan content shown by DMMB staining of cell pellets after chondrogenic (A) and hypertrophic conditions (B), as well as different collagen 2 production as revealed by immunohistochemistry against collagen 2 of cell pellets after chondrogenic (C) and hypertrophic conditions (D). Enhancement of hypertrophy shown by immunohistochemistry against collagen 10 between chondrogenic (E) and hypertrophic (F) conditioned cell pellets as well as by ALP staining of chondrogenic (G) and hypertrophic (H) conditioned cell pellets (Scale bar=500 μm).
|
TGFβ signaling activity is reduced under hypertrophic conditions
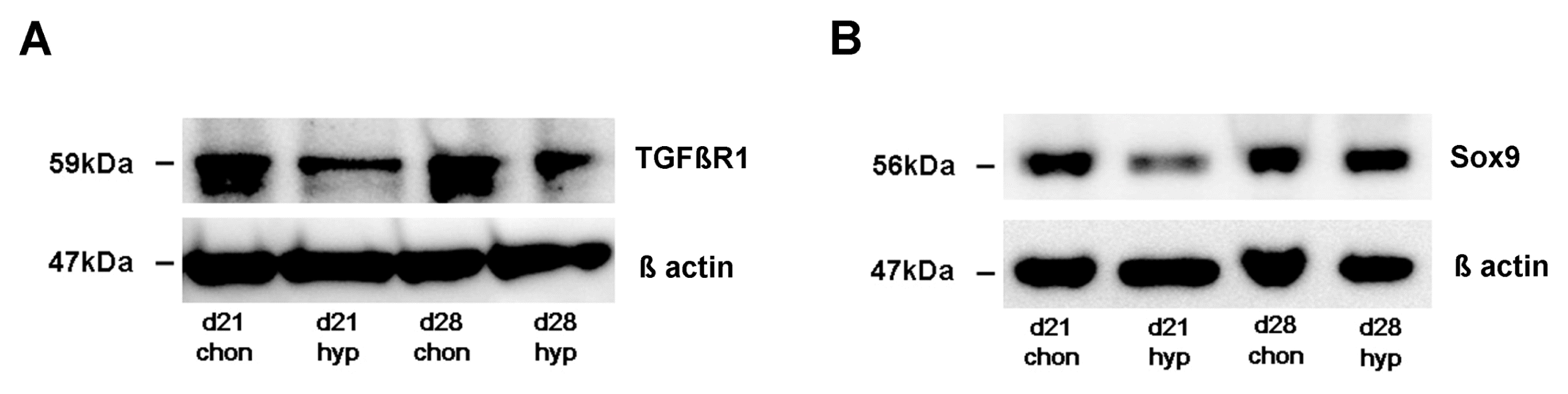
We detected a significant down-regulation of TGFβ receptor expression under hypertrophy enhancing conditions. Real time PCR revealed that the TGFβ receptor 1 (TGFβR1) is significantly down-regulated under hypertrophic conditions on day 17, day 21 and day 28 compared to chondrogenic conditions (Fig. 2A). The TGFβ receptor 2 (TGFβR2) is significantly down-regulated on day 28 in hypertrophic MSC pellets (Fig. 2B). On protein level, Western Blot analysis detected a decreased amount of TGFβR1 protein under hypertrophic conditions on day 21 and day 28 compared to chondrogenic conditions (Fig. 4A). The amount of Sox9 protein is reduced on day 21 under hypertrophic conditions but not on day 28 (Fig. 4B). Western blotting thus reinforced our rtPCR results and revealed again suppression of TGFβ receptors 1 and 2 expression as well as Sox 9 release under hypertrophic conditions.
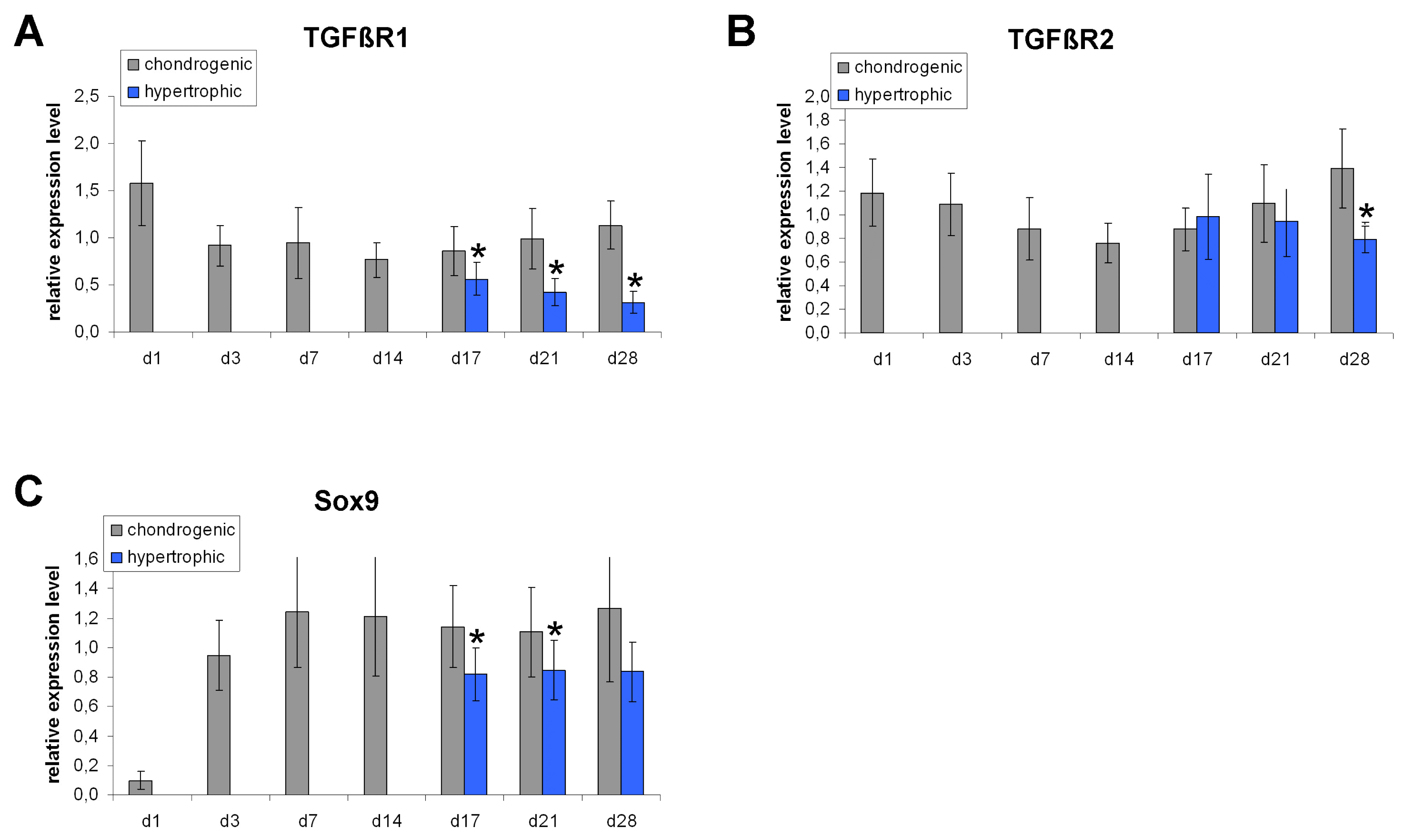 | Fig. 2Gene expression analysis of TGFβR1, TGFβR2 and Sox9 normalized to HPRT in MSC pellet cultures under chondrogenic and hypertrophy enhancing conditions analyzed by real time PCR. TGFβR1 is significantly down-regulated under hypertrophic conditions on day 17, 21 and 28 (A). TGFβR2 is down-regulated under hypertrophic conditions on day 28 (B). Sox9 is down-regulated on day 17 and 21 in hypertrophic MSC pellets (C). n=7 different donors.
|
In order to investigate, whether there are differences in TGFβ signaling activity between chondrogenic and hypertrophic MSC pellets, we performed Western Blot analysis for the phosphorylated forms of Smad2 and Smad3. The amount of phospho-Smad2 and phospho-Smad3 is clearly reduced in hypertrophic MSC pellets on day 21 and day 28 compared to chondrogenic pellets. The total amount of Smad2 and Smad3 protein and β actin were used as loading control (Fig. 3).
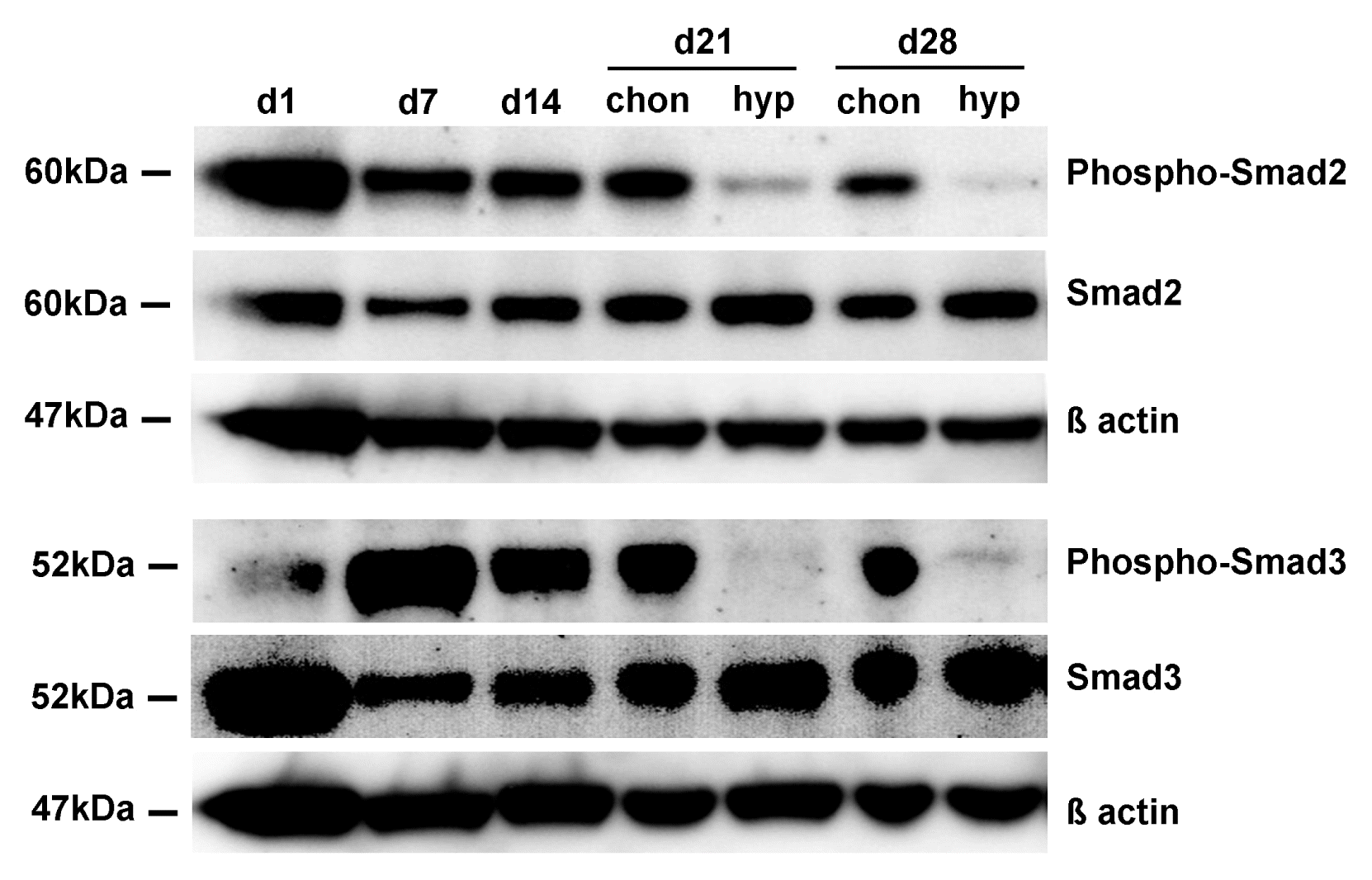 | Fig. 3TGFβ signaling activity. Western Blot analysis of phospho-Smad2 and phospho-Smad3 under chondrogenic and hypertrophic conditions. The amount of phospho-Smad2 and phospho-Smad3 is reduced under hypertrophic conditions on day 21 and day 28 compared to chondrogenic conditions. The total amount of Smad2/Smad3 and β actin were taken as loading control.
|
TGFβ signaling associated genes are down-regulated under hypertrophic conditions
In order to elucidate a possible role of TGFβ signaling in the regulation of MSC hypertrophy we compared the expression of TGFβ signaling associated genes between chondrogenic and hypertrophic conditions. The TGFβ signaling associated transcription factor (via Smad 2/3) Sox9 is significantly down-regulated on day 17 and day 21 under hypertrophy enhancing conditions. On day 28 there is a tendency towards decreased Sox9 expression in the hypertrophic group, without reaching statistical significance (Fig. 2C).
The TGFβ-receptor-type-1 inhibitor SB431542
We investigated the effect of the TGFβ antagonist SB431542 on hypertrophic differentiation in the in vitro hMSC hypertrophy model. Under chondrogenic conditions treatment with SB431542 does not enhance hypertrophy. There is no histological difference between MSC pellets that were treated with low doses of SB431542 (Fig. 5B, C) and chondrogenic control aggregates (Fig. 5A). Under all conditions a hyaline cartilage like morphology with weak hallmarks of hypertrophy developed. In contrast, high doses of the TGFβ receptor inhibitor lead to a dedifferentiation of the pellets (Fig. 5D). Similarly, no difference in ALP staining can be detected between chondrogenic control aggregates (Fig. 5E) and SB431542 treated aggregates (Fig. 5F~H). Collagen type II staining is unchanged between chondrogenic control pellets (Fig. 5I) and MSC pellets that were treated with low doses of SB431542 (Fig. 5J, K). However, high doses of the TGFβ inhibitor strongly reduce Collagen type II staining (Fig. 5L). Collagen type X staining is weak in chondrogenic control aggregates (Fig. 5M) and in aggregates that were treated with low doses of SB431542 (Fig. 5N, O). In MSC pellets that were treated with 10 μM of the TGFβ inhibitor, no collagen type X staining can be detected (Fig. 5P).
 | Fig. 5Histological appearance of MSC pellet cultures on day 28 after SB431542 treatment under chondrogenic conditions. No change in DMMB, ALP, collagen type II and collagen type X staining can be detected between chondrogenic control pellets (A, E, I, M) and pellets treated with low doses of SB431542 (B, C, F, G, J, K, N, O). High doses of SB431542 lead to a dedifferentiation of the cells shown by decreased DMMB staining (D) and collagen type II staining (L) (Scale bar=200 μm).
|
In the following, we aimed to investigate whether we can increase hypertrophy under chondrogenic conditions by simultaniously inhibiting TGFβ signaling with SB431542 and addition of bone morphogenetic protein 4 (BMP4). The sole/exclusive addition of BMP4 to chondrogenic medium does not increase the number of hypertrophic cells (Fig. 6A). Simultaneous addition of BMP4 and the TGFβ inhibitor SB431542 increases the number of hypertrophic cells dose dependently with increasing SB431542 concentration under chondrogenic conditions (Fig. 6B~D).
 | Fig. 6Histological appearance of MSC pellet cultures on day 28 after SB431542 and BMP4 treatment under chondrogenic conditions. Concomitant application of SB431542 and BMP4 increases hypertrophy in a dose dependent manner shown by an increased amount of hypertrophic (B~D) and ALP positive (F~H) cells and increased collagen type X staining (N~P) after SB431542 and BMP4 treatment, compared to BMP4 only treatment (A, E, M). No difference in collagen type II staining can be detected between BMP4 treated control pellets (I) and pellets treated with SB431542 and BMP4 (J~L) (Scale bar=200 μm).
|
Similarly, there are only few ALP positive cells in chondrogenic MSC pellets that were only treated with BMP4 (Fig. 6E). The amount of ALP positive cells increases in chondrogenic MSC pellets that were treated with both, BMP4 and the TGFβ inhibitor (Fig. 6F~H). The highest amount of ALP positive cells can be detected in pellets that were treated with BMP4 and 10 μM SB431542 (Fig. 6H). There is no difference in Collagen type II staining between chondrogenic MSC pellets that were treated with BMP4 (Fig. 6I) and chondrogenic MSC pellets that were treated with BMP4 and the TGFβ inhibitor (Fig. 6J~L). MSC pellets that were treated with the highest concentration of the TGFβ inhibitor SB431542 together with BMP4 do not dedifferentiate and show a strong collagen type II staining, indicating that BMP4 preserves chondrogenic differentiation after SB431542 treatment (Fig. 6L). Collagen type X staining is low in chondrogenic MSC pellets that were treated with BMP4 (Fig. 6M). Treatment with BMP4 and SB431542 increases collagen type X staining (Fig. 6N~P) with the highest effects to be shown for lower SB431542 concentrations. However higher concentrations of SB431542 e.g. 3μM and 10μM seem to mitigate the production of collagen type X within the pellets observed with lower doses e.g. 0.5μM of SB431542.
Under hypertrophic conditions, the TGFβ inhibitor SB431542 does not further enhance hypertrophy. Nevertheless there are differences detectable between hypertrophic control pellets (Fig. 7A) and pellets that were treated additionally with SB431542 (Fig. 7B~D). In a dose dependent manner cells treated with SB431542 under hypertrophic challenge reveal smaller cells with smallest cell sizes observable in the group treated with 0.5μM of TGFβ-receptor-type-1 inhibitor SB431542 whereas large lacunae are visible in the group treated with 10μM of TGFβ-receptor-type-1 inhibitor SB431542 – the highest dosage in the present study. After all, cellse still maintain the chondrogenic – but hypertrophic – phenotype, despite increased inhibition of TGF-β signaling. Pro-hypertrophic signaling is also still evident in the SB431542-treated groups as the amount of ALP positive cells is unchanged between hypertrophic control aggregates (Fig. 7E) and SB431542 treated hypertrophic aggregates (Fig. 7F~H). No difference in collagen type II staining can be detected between the hypertrophic group (Fig. 7I) and the SB431542 treated hypertrophic group (Fig. 7J~L). Under hypertrophic control conditions, collagen type X staining is strong (Fig. 7M). Treatment with 0,5 μM and 3 μM SB431542 does not change collagen X staining (Fig. 7N, O). However, treatment with 10 μM SB431542 and BMP 4 slightly reduces collagen type X staining (Fig. 7P).
 | Fig. 7Histological appearance of MSC pellet cultures on day 28 after SB431542 treatment under hypertrophic conditions. No difference in DMMB, ALP, collagen type II and collagen type X staining can be detected between hypertrophic control pellets (A, E, I, M) and SB431542 treated pellets (B~D, F~H, J~L, N~P) (Scale bar=200 μm).
|
Go to :
Discussion
As previously reported (10) hypertrophic conditions were achieved by addition of T3 and withdrawal of dexamethasone after chondrogenic predifferentiation. Similar findings of the present investigation mirror the characteristics of hypertrophic chondrocytes found in the growth plate (7, 10, 19), allowing to use this hypertrophy model for further analysis.
Furthermore, TGFβ downstream signaling activity is reduced under hypertrophic conditions. Our findings suggest that this effect is most likely a result of downregulated TGFβ Receptor expression. These findings are in accordance with a previous study by Serra et al., that showed that transgenic mice, which express a defective TGFβ Receptor type 2 (TGFβR2), have increased hypertrophic differentiation and premature collagen type X expression in the growth plate (23). The exact mechanism by which hypertrophic conditions reduce the expression of TGFβ receptors remains to be elucidated. However, our results suggest that the downregulation of TGFβ-receptors contribute to hypertrophy in consonance with reduced TGFβ ligands. TGFβR1 was shown to be present mainly in resting and hypertrophic chondrocytes within the growth plate and in the perichondrium (25). Further on in situ hybrdization of TGF-β receptor expression during development suggested that TGFβR2 is expressed in chondrogenic condensation centers in embryonal chicken limb buds (26), but not TGFβR1. In a mouse model TGF βR2 was detected in developing joints especially in inter-zone cells (27). In addition to that Spagnoli et al. reported, that TGFβ2R conditioned knockouts failed to develop proximal and medial interphalangeal joints, displayed smaller ossification centers and therefore showed reduced limb length (27). After all it has to be mentioned, that complete knockouts of TGFβR2 are nonviable.
TGFβ signaling utilizes intracellular Smad proteins to transduce the signal from the activated receptor to the nucleus. Upon activation of the TGFβ receptor, intracellular Smads2 and Smads3 get phosphorylated and move to the nucleus to activate downstream gene expression. The results of our performed Western Blot analysis for the phosphorylated forms of Smad2 and Smad3 indicate that TGFβ signaling is less active under hypertrophic conditions. TGFβ signaling is follows a strict spatio-temporal expression pattern in vivo during enchondral ossification (28) and is downregulated in hypertrophic areas of the growth plate (28). These findinings have been reported in mice (28) and rats (29). Our results show that human hypertrophic MSCs show similar expression patterns of TGF β in vitro and therefore it is likely that the above in vivo results may be transferred to human MSC chondrogenesis. Also, Western blotting reinforced our rtPCR results and revealed again suppression of TGFβ receptors 1 and 2 as well as Sox 9 under hypertrophic conditions. Sox 9 is known to inhibit hypertrophy by inhibiting Runx2 (30) via Nkx3.2 (31). The main reason for downregulation of Sox 9 in the context of hypertrophy is the downregulation of Smad 2/3 phosphorylation due to reduced TGFβ receptor signaling, orchestrated by lack of the ligand – TGF β – and reduction of TGFβR1 and TGFβR2 expression. Our study shows that TGFβ associated genes are downregulated in hypertrophic conditions.
The experimental data obtained from gene expression analysis and Western blotting showed that TGFβ receptors are down-regulated and TGFβ signaling activity is reduced under hypertrophic conditions. We hypothesized that the reduced TGFβ signaling activity under hypertrophic conditions contributes to the enhancement of hypertrophy. In order to test this, we investigated the effect of the TGFβ antagonist SB431542 on hypertrophic differentiation in the in vitro hMSC hypertrophy model. SB431542 inhibits TGFβR1 kinase activity and thereby inhibits the phosphorylation of Smad3 (32, 33). The inhibition of TGFβ signaling with SB431542 does not enhance hypertrophy under chondrogenic conditions. During endochondral ossification loss of function models of TGF β, leading to decreased TGFβ signaling, increased chondrocyte hypertrophy in vivo (23, 24). High doses of the TGFβ inhibitor lead to a dedifferentiation of the cells shown by decreased metachromatic DMMB staining and decreased collagen type II staining, suggesting a dose dependent inhibition of TGFβ signaling.
Previous results showed that bone morphogenetic protein 4 (BMP4) has strong pro-hypertrophic properties and is able to increase hypertrophy under hypertrophic but not under chondrogenic conditions (34). Adding BMP4 under chrondrogenic conditions presented similar results in this investigation.
We were able to show that the simultaneous addition of the TGFβ inhibitor and BMP4 increases hypertrophy in hMSCs. This was shown by an increased amount of hypertrophic, ALP positive cells and increased collagen type X staining. These findings in human MSCs are in accordance with reports by Callahan et al. (33), who found in bone organ cultures of mouse embryos a prohypertrophic effect, when blocking endogenous TGF-β stimulation by addition of SB431542 under simultaneous BMP-2 treatment. Further on it was suggested that TGF-β signaling induces a negative feedback loop on BMP signaling by Ski related novel protein N (SnoN), which is reduced upon addition of SB431542 (35). We were able to show that the simultaneous addition of the TGFβ inhibitor and BMP4 increases hypertrophy in hMSCs. This was proven by an increased amount of hypertrophic, ALP positive cells and increased collagen type X staining.
Despite the inhibition of TGFβ sginaling with a high dosage of SB431542, hypertrophy could not be enhanced which gives evidence for the maintainance of the chondrogenic phenotype. This finding suggests that alternative chondrogenic stimulation e.g. via the BMP-signaling pathway is capable of maintaining this differentiation status in vitro. Controversially, loss of function models of TGF β, leading to decreased TGFβ signaling, increased chondrocyte hypertrophy in vivo (22, 23) during endochondral ossification. The collagen II and X staining under treatment with SB431542 and BMP 4 mirrors the results of SB431542 treatment under purely chondrogenic conditions. Of note is that the cell phenotype was converted dose dependently towards hypertrophy under the regimen off the two prohypertrophic agents BMP 4 (34) and the TGFβ-receptor-type-1 inhibitor SB431542 (32) while hypertrophy in terms of loss of collagen 2 staining and increased collagen 10 production is not evident.
In summary, our results show that TGFβ signaling is downregulated under hypertrophy enhancing conditions using an established in vitro hypertrophy model in human MSCs. We could show lower TGFβ receptor expression, reduced Smad-2 and 3 activation and lower expression of downstream molecules. Further the TGFβ inhibitor SB-431542 increases hypertrophy under chondrogenic conditions when applied together with BMP4. However SB431542 is not capable to enhance hypertrophy under chondrogenic conditions without additional BMP-4 treatment. Together with previous findings, these results suggest that the enhancement of hypertrophy in this model is a result of both, activation of pro-hypertrophic BMP signalling (32) and reduction of anti-hypertrophic TGFβ signaling. Thus similar regulatory mechanisms of hypertrophy as in embryonic endochondral ossification plays a role in this in vitro model.
Go to :
Conclusions
These experiments showed that the expression of TGF β-type-1- and 2-receptor and Sox 9 were significantly downregulated upon hypertrophic challenge of the cell constructs as well as phosphorylation of downstream regulators such as Smad 2 and 3 was reduced. Inhibition of TGFβ-receptor-type-1 by SB431542 did not initiate hypertrophy under pro-chondrogenic conditions, but enhanced hypertrophic hallmarks when applied simultaneously with BMP-4.
Overall our results suggest that hypertrophy in this model is enhanced as a result of both activation of pro-hypertrophic BMP signaling and reduction of anti-hypertrophic TGFβ signaling.
Go to :
 XML Download
XML Download