

 PDF
PDF Citation
Citation Print
Print


Introduction
Acute renal failure or acute kidney injury (AKI) with high morbidity and mortality rate (30~70%) is a rapid decline in renal functions, which is to excrete nitrogenous waste products and to maintain electrolyte and fluid balance. The major pathophysiological consequence is the loss of tubular epithelial cells (1). Persistent AKI can become a life-threatening clinical condition. In AKI, the mechanism is a complex interplay among vascular, tubular, and inflammatory factors followed to either repair process to restore epithelial differentiation or incomplete recovery may result in progressive chronic kidney disease (CKD). Effective therapeutic treatments are unavailable. Therefore, various stem cells induced regenerative preclinical therapies have been reported (2). In a variety of undifferentiated adult cells, ex vivo expansion of adipose-derived mesenchymal stem cells (Ad-MSCs) is considered to be less invasive with ease of collection, higher proliferation rate, and preparation for autologous or allogeneic transplantations. MSCs are defined by their adherence to plastic in culture and multipotential capacity. It may provide external support along with in vivo stem cells as well as may play a role towards differentiation and replacement of damaged cells as a cellular therapeutic approach (3). The multitude of trophic factors includes growth factors, cytokines, and polypeptides secreted by stem cells can produce paracrine and endocrine effects to regulate epithelial proliferation, cell differentiation, and to modulate inflammation (4, 5). Although the exact detailed mechanism is still not fractioned, these factors may activate the regeneration process. The aim of the current study is to investigate the effects of the mouse adipose tissue-derived mesenchymal stem cells (mAd-MSCs) transplantation for regeneration and amelioration of AKI with recovered structure and function for the survival of mice. Further, it was taken an account to explore the mechanism of renal parenchymal changes and proliferation of injured renal cells in response to allogeneic transplantation of mAd-MSCs in balb/c mice with severe renal toxicity caused by cisplatin (CP). CP was used to induce an acute renal failure condition in balb/c mice, which have been widely and effectively used as a chemotherapeutic agent. CP accumulates in tubular epithelial cells at the S3 segment of proximal convoluted tubules (PCT). In this study, we have followed Risk, Injury, Failure, Loss, End-stage renal disease (RIFLE) classification and AKI network (AKIN) staging systems for the development of the CP-induced renal toxicity model (6, 7). Here, we have been connecting the biochemical and genetic mechanism of reconstruction and proliferation of tubules for improvement in the renal functions and consequently, the survival of the mice. It demonstrated the successful promotion of injured renal cellular replacement and tissue remodeling via optimal delivery of Ad-MSCs infusion, and it would be one of the best clinical sources in regenerative medicine.
Materials and Methods
mAd-MSCs isolation and culture
Inguinal adipose tissues of Balb/c mice were isolated in Hank’s Balanced Salt Solution (HBSS, cat.no.14025092, Life Technologies, USA) and washed twice with HBSS at 280 g to remove blood and liquid. Clear fat’s tissues were cut into 3 mm small fragments and incubated in the solution of HBSS contained 100 units/ml collagenase type I (cat.no.17100-017, Life Technologies, USA) along with 3 mM CaCl2 for two hours at 37°C and 5% CO2. After neutralization with fetal bovine serum (FBS, ref.no.16000-044, Gibco Life Technologies, USA), cells were centrifuged, the pellet was washed in HBSS, and filtered through 70 μm then 40 μm nylon mesh. mAd-MSCs were cultured in Minimum Essential Medium Eagle-Alpha Modification (α-MEM, cat.no.32561-037, Life Technologies, USA) containing 100 U/ml penicillin/streptomycin and 10% FBS in 25 cm2 flasks. The flasks were incubated at 37°C in the humid atmosphere at 5% CO2. Non-adherent cells were removed by medium replacement after every 2~3 days. Upon confluence at 70% density, cells were sub-cultured through 1X TrypLE select (cat.no.12563-011, Gibco Life Technologies, USA) in 1:3 ratio. Passage 2 mAd-MSCs were used for transplantation studies in AKI model (8).
Characterization of mAd-MSCs
mAd-MSCs were first analyzed by transcriptomic expressions of CD90 (Thy1) and CD44 (HCAM) (H-300) by RT-PCR. The probes are mentioned in Supplementary Table S1. Further, it was confirmed by positive expressions of these markers and negative expressions of CD45 (H-230) and CD34 (C-18), through immunocytochemistry (ICC). For ICC mAd-MSCs were cultured in 4 well chambered slides and fixed for 2 minutes in 4% paraformaldehyde, washed with Ca+, Mg+ PBS, and blocked with goat serum for an hour at room temperature. After being washed with PBS, the cells were incubated at 1:50 concentrations of rabbit polyclonal primary antibodies of CD90 (sc-9163), CD44 (sc-7946), CD45 (sc-25590), and CD34 (sc-7045) overnight at 4°C. Next day, after washing, goat anti-rabbit IgG-fluorescein isothiocyanate (FIT-C)-conjugated secondary antibody (sc-2012) was placed for an hour in dark at room temperature. 4′, 6-diamidino-2-phenylindole (DAPI; sc3598; 0.5 μg/ml) was used to stain nuclei. All the antibodies and DAPI were purchased from Santa Cruz Biotechnology. Inc., USA. The mAd-MSC phenotype and multipotency were further confirmed by the ability to differentiate into osteocytes and adipocytes. Osteogenesis (cat.no.SCR028, Australia) was carried out in inductive media containing 1 mM dexamethasone on vitronectin/collagen coated 24 well plates according to the manufacturer’s instructions. After 20 days, Alizarin Red S dye, and Von Kossa stains were used to observe calcified differentiated cells. For adipogenesis 100 nM dexamethasone was used in inductive media (cat.no.TM005, ABM, Canada) with 50 mg/ml indomethacin. After 21 days, differentiated cells were observed through Oil red O staining. Microscopic analyses were carried out at 20~40× magnifications.
Study design and development of AKI
The experimental animal procedures were approved by the ethical and the animal care and use committee of Sindh Institute of Urology and Transplantation (SIUT)-Karachi, according to international standards. 6~8 week old male Balb/c mice (25~30 g; n=70, 10/group) were maintained under pathogen-free conditions. They received standard food pellets and water ad libitum 12 hours light and 12 hours dark cycle, 22±2°C temperature, and 40~45% humidity. Mice were divided into seven groups according to the duration (days) post CP injection and harvesting of samples. The groups included were (i) Normal/Control; (ii) 24 hr; (iii) 48 hr; (iv) 72 hr; (v) 96 hr; (vi) 120 hr; (vii) CP+mAd-MSCs transplanted. AKI was induced by single subcutaneous infusion of 18 mg/kg Cisplatin (CP) (cat.no.3410101336-20504, Korea United Pharm. Inc., Korea) in physiological saline and control group received normal saline instead. Mice weighed daily and euthanized by anesthetic overdose at each time point of the experimental period. Blood serum and kidney tissues were collected between 24~120 hours of CP injection from CP groups, and CP+mAd-MSCs transplanted mice were euthanized after a week (total 12 days). Weight (gram) and survival of mice were observed.
Cell preparation, fluorescent dyes labeling, and transplantation of mAd-MSCs
For single cell preparation, 90~95% viable mAd-MSCs were harvested using accutase (cat.no.A1110501, Fisher Scientific, Norway), washed thoroughly, and suspended in PBS in presence of antithrombin Eskinase (Streptokinase B.P, 1.5MIU, Pakistan). Cells were suspended in 700 μl 1X PBS contained 3 μl (0.0045MIU) Eskinase (~0.00130/200 μl PBS) and gently filtered through 70 μm then 40 μm nylon mesh. Prior to this, 1 ml syringes were triturated through heparin (HeptaRotex; 5,000 units/ml, Germany) and sodium bicarbonate (7.5%) (REXA-BiCARB, Pakistan). mAd-MSCs were filled in the syringe through 200 μl tips and kept at 37°C until transplantation within ten minutes. For in vivo tracking of cells within 24~72 hours fluorescence labeling dyes, 5-chloromethylfluorescein diacetate (Cell Tracker®CMFDA, cat.no.C7025, Life Technologies, USA) (Ex.492/Em.517 nm) and Vybrant Dil (cat.no.V-22885 Molecular Probes, Inc., USA) (Ex.549/Em.565) (50 μg/ml) were standardized to mAd-MSCs before infusion. mAd-MSCs were administered to AKI mice after 72 hours of injury (group vii: CP+mAd-MSCs transplanted mice). In different experiments, 2.5×107 cells/kg (0.5~1×106/200 μl) body weight labeled and unlabeled cells were infused gently in the lateral tail vein of CP-treated Balb/c mice.
Histopathology
Harvested kidneys were rinsed in saline (0.9% sodium chloride) and perfused in 10% neutral buffered formalin. After appropriate dehydration, kidney slices were embedded in paraffin, deparaffinized with xylene, rehydrated, and sectioned at 4 μm thick slices using a microtome. It proceeded to stain via Hematoxylin and Eosin (H&E), Periodic Acid-Schiff’s staining (PAS), and Masson’s Trichrome staining (TRI). Corticomedullary fields were recorded from each kidney section by the light microscope (Nikon Eclipse Ti-S) equipped with a DS-L3 (DS-Fi2) camera. Tubular damage was identified by the loss of brush border, lumen dilatation or collapse, vacuolation, and desquamation of epithelial cells in renal tubules after CP treatment. Histopathology grading by light microscopy was considered for renal injury. The grades were given to tubular necrosis that loss of brush border or tubular epithelial cells, protein cast formation, tubular dilatation, and accumulation of inflammatory infiltrate in ten randomly chosen non-overlapping fields (40× Magnification) of kidney sections. The scores for grading were based on the percentage of the affected parameter, which is as follows: 0 (no change/normal architecture), 1 (minimal changes ≤ 15%), 2 (mild changes 15≤25%), 3 (moderate degeneration 25~50%), 4 (severe degeneration 50~75%), and 5 (extremely severe degeneration ≥75~100%) (9).
Immunohistochemistry
To assess morphology, kidney specimens were snap-frozen at −20°C in optimum cutting temperature (O.C.T.) solution. For detection of proliferating cells in kidney sections, 5-Bromo-2′-Deoxyuridine+5-Fluoro-2′ Deoxyuridine (BrdU; 00-0103) was intraperitoneally injected (100 mg/kg) in mice for an hour before being sacrificed. AlexaFluor®BrdU-Mouse Monoclonal (Clone MoBU-1; Invitrogen) antibody (Ex.495/Em.519 nm) was used in 1:500 dilution in the sections, which were already transplanted with Vybrant Dil (Ex.549/Em.565 nm) labeled mAd-MSCs. The kidney sections were adsorbed on polylysine-coated glass slides, air-dried, de-paraffinized, and then placed in a water bath. The sections were treated with heating for antigen retrieval. For peroxidase-linked immunostaining, endogenous peroxidase was removed by 0.05 M hydrogen peroxide (H2O2) for five minutes. Non-specificities were blocked by 5% bovine serum albumin. The kidney specimens were incubated with rabbit polyclonal Megalin (sc25470, Santa Cruz Biotechnology. Inc., USA) and Kidney Injury Molecule 1 (Kim-1) (ab47635, abcam, UK) against mouse at 1:1000 and 1:1500 dilutions respectively. Biotinylated horse (DSP-999) and Biotinylated goat (DGP-999) (Spring Bioscience Corp., USA) anti-polyvalent horseradish peroxidase (HRP) secondaries were used against Megalin and Kim-1 primary antibodies respectively. 3, 3′-diaminobenzidine (DAB) was used as a chromogen. The sections were counterstained with hematoxylin and mounted with DPX (cat.no.4458, SIGMA). Fluorescence microscope (Nikon Eclipse Ti-S) was used to acquire images at 20~40× magnifications.
Functional analyses
Serum creatinine (sCr) (cat.no.CR510), blood urea (cat.no.UR107), and total urinary protein (cat.no.UP 1570) were measured in aforementioned groups. These kits were purchased from Randox Laboratories Limited, UK. Change in biochemical parameters was considered for renal functions’s assessment and the survival of mice.
PCR and TaqMan qRT-PCR
Renal tissues were homogenized, sonicated, and total RNA was isolated using Ambion kit (cat.no.12183018A, PureLink TM RNA Mini Kit, Invitrogen, Life Technologies, USA) for the biological and technical triplicates and 0.5 μg RNA was used to synthesized cDNA according to the manufacturer’s instructions (part no.4368813, High Capacity cDNA Revert Transcriptase Kit, Applied Biosystems Inc., USA). It was amplified under the following PCR conditions: 94°C for five minutes, 30 cycles of PCR (94°C for 30 sec, 56°C for 30 sec, 72°C for 30 sec) 72°C for seven minutes and then at 4°C using Master Mix (cat.no.10572-063, PCR Supermix, Invitrogen, USA). Samples were electrophoresed at 2% agarose gel. For a standard reaction in real-time PCR, cDNA in 1:10 dilution was used as a PCR template. cDNA was mixed together with 2.5 nM of each primer in a final volume of 25 μl. Real-time PCR was performed using LightCycler TaqMan Master Mix (Roche, Germany) through manufacturer’s guidelines. An endogenous ‘housekeeping’ gene, 18S rRNA was quantified and used to normalize the results. All the genes were compared with control (normal adult mice) and relative genes expressions were quantified through 2^−ΔΔCt method. The probe sequences are given in Supplemental Table 1.
Statistical analyses
Quantitative data were expressed as means±s.e.m., or means±s.d. ANOVA was performed, followed to Post Hoc Tucky’s correction (SPSS 20.0 for Windows, SPSS Inc., USA; or MedCalc version 17.9.5) to compare multiple groups. Kaplan-Meier test was performed for survival function analysis. 95% confidence interval (*p≤0.05) was considered statistically significant.
Results
mAd-MSC phenotype and characterization
mAd-MSCs were shown to be adherent to the plastic surface, spindle-shaped, and fibroblast appearance with colony forming properties, which were observed through light microscopy. mAd-MSCs rarely expressed CD45 and CD34 expressions, which are specific surface markers for hematopoietic cells while CD90 and CD44 are positive markers of stem cells, and were also positively detected by RT-PCR and ICC. Osteogenic and adipogenic differentiation assays confirmed the multipotential capacity of isolated mAd-MSCs by the formation of hydroxyapatite minerals and fatty depots respectively (Fig. 1).
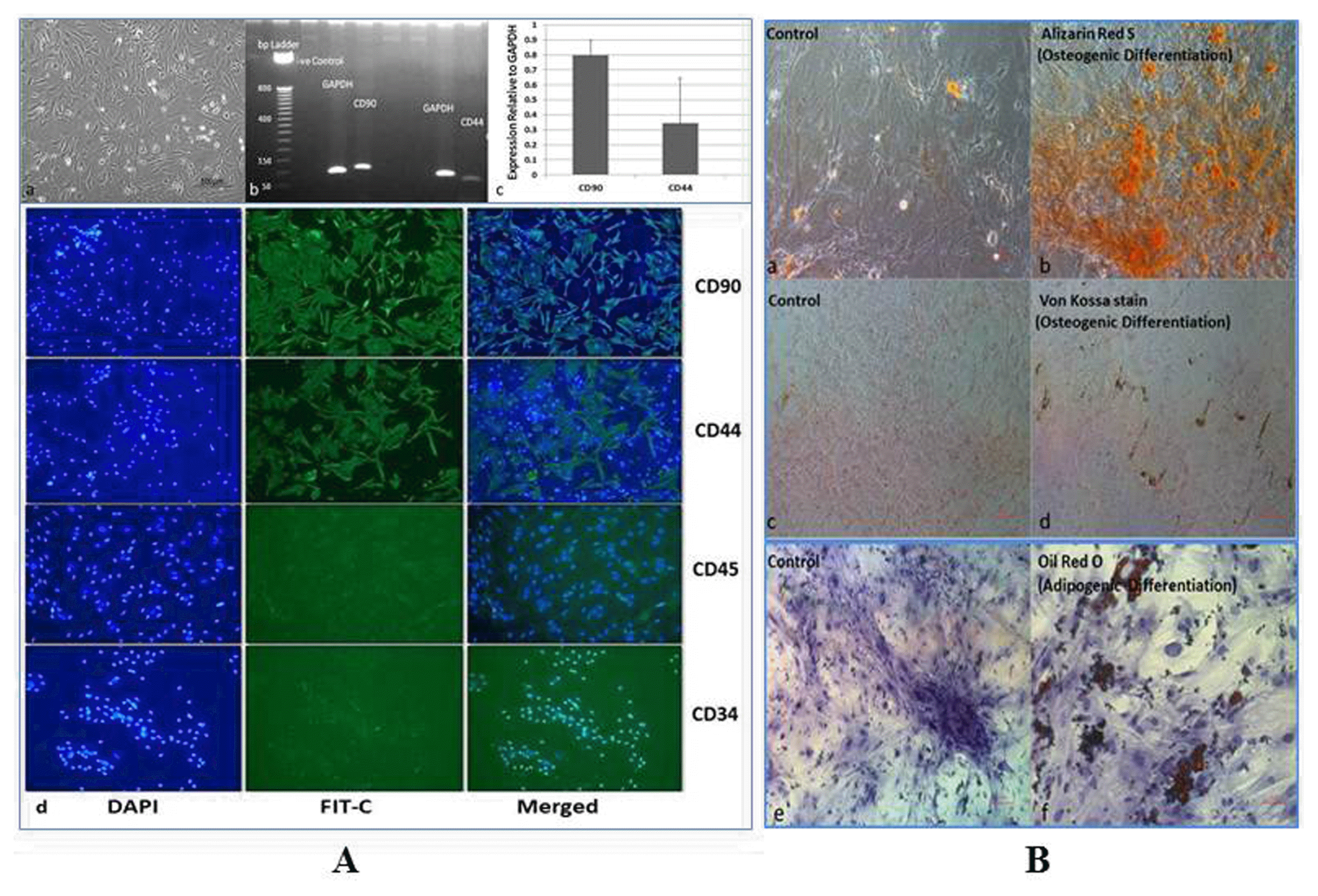
Fig. 1
Morphometry and characterization of mAd-MSCs from Balb/c mice. (A) (a) mAD-MSCs with spindle-shaped morphology (b) The transcriptomic expressions of stem cells markers CD90 and CD44 on 2% agarose gel electrophoresis and (c) Semi-quantification of these markers relative to Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) (d) Immunocytochemistry for positive expressions of stem cells markers CD90 and CD44 (raw1 & 2), and negative expressions of CD45 and CD34 (raw 3 & 4). All proteins are shown with 4′, 6-diamidino-2-phenylindole (DAPI), fluorescein isothiocyanate (FIT-C) conjugated secondary antibody and merged images (20× Magnification). (B) (a~d) In vitro differentiation of mAd-MSCs into osteogenic lineage by the formation of calcium hydroxyapatite mineral positive cells, which were visualized by Alizarin Red S (orange-red) (b) and Von Kossa stain (brown to black pigments) (d) both are shown with respective controls (a & c). (f) Adipogenic lineage differentiation of mAd-MSCs for the formation of intracellular lipid vacuoles, which were visualized by Oil Red O stain and is shown with its control (e) (40× Magnification).
![]()
In vivo mAd-MSCs accumulation, engraftment, and extent of proliferation in severely injured kidneys
Administered cells were detected by fluorescence microscopy in kidney sections. mAd-MSCs were identified by CMFDA (green) and Dil (red) labeled cells in the renal parenchyma within 24~72 hours following systemic administration of 0.5~1×106 mAd-MSCs. It demonstrates localization and engraftment potential of mAd-MSCs under injury cues. Most cells were localized in the cortex and outer medulla in the region of proximal tubular cells; the most prominent region of CP-induced renal injury. mAd-MSCs were not observed in the heart, some observed in the liver, and many cells were observed in the lungs (Fig. 2A~C). Nuclear stains via anti-BrdU displayed proliferating tubular epithelial cells in the cortical region as compared to control and other body organs of the same mouse. BrdU is incorporated into newly synthesized DNA during the S phase of the cell cycle, which demonstrates enhanced multiplication or proliferation of cells in renal tubules. Most glomeruli contained a few BrdU-retained cells (Fig. 2C, D).
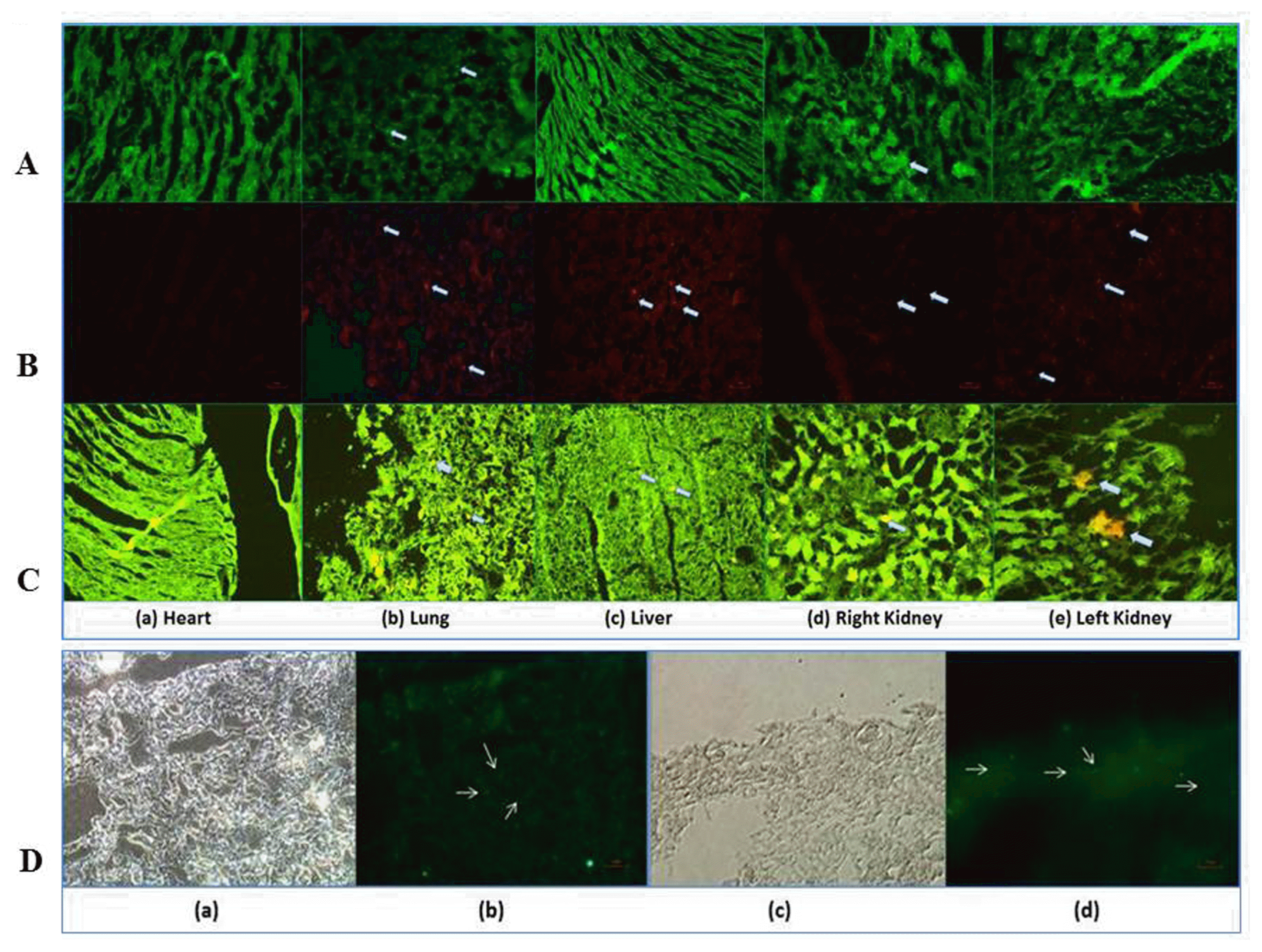
Fig. 2
mAd-MSCs localized to the kidney within 24~72 hours, which facilitate the regeneration of renal tubular cells by excessive proliferation rate. Fluorescence microscopic examinations of heart, lung, liver, and kidney are demonstrated for in vivo tracking or homing of mAd-MSCs within 24~72 hours in AKI mice and proliferation of injured kidney tissues post seven days of intravenous mAd-MSCs infusion in CP-treated mice. The viability and proliferation of cells were not affected by the dyes like CMFDA and Dil. In vivo transplanted mAd-MSCs were observed in kidneys of AKI produced by CP (18 mg/kg, s.c.). Localization of cells is shown by arrows. (A) CMFDA labeled mAd-MSCs in CP-induced AKI (B) Dil labeled mAd-MSCs in CP-induced AKI. (C) Merged images of Co-stained Dil labeled mAd-MSCs and fluorescein isothiocyanate (FIT-C) labeled anti-BrdU staining following injection of BrdU reagent to assess proliferation of cells in injured kidney slices. In each panel (a) Heart (b) Lung (c) Liver (d) Right Kidney (e) Left Kidney (20× Magnification). (D) Micrograph of the kidney slices of FIT-C labeled anti-BrdU staining. (a & b) CP-treated mice (96 hrs CP), (c & d) CP+mAd-MSCs transplanted kidney sections (20× Magnification).
![]()
Histological and immunohistological impact of CP and mAd-MSCs transplantation in CP-induced AKI
H&E, PAS staining, and histopathological grading displayed significant (***p≤0.001) severe tubular cell necrosis, brush border cells loss, and inflammatory infiltrates with luminal casts formation in animals following exposure to CP (18 mg/kg, s.c.) from 24~120 hours as compared to the normal architecture of renal cells in control mice. After 24 hours, the main features were vacuolization in tubules, sloughing of cells, and removal of nuclei from tubules while other organs, heart, lung, and liver remain in normal architecture (data not shown). Most of the renal parenchyma was indistinguishable. Atrophied cells and the injuries were found to be more severe after 72~96 hours of CP, and mortality was high between 96~120 hours. As a result of these changes, segmental hyalinization was observed in the cortical region. Glomeruli were also affected; atrophied, segmental shrinkage in Bowmen’s capsules were observed in 96 hours of CP-treated sections. H&E staining showed that congestion was started from 24 hours of CP infusion, and low congestion was found in the group that received mAd-MSCs. Prominent casts were observed after 48 hours of CP infusion while hexagonal or rectangular shape tubular dilatations were observed after 96 hours of CP infusion. Histopathology also revealed the accumulation of nuclei after patchy or diffused denudation and cellular blabbing of renal epithelial cells post 96 hours of CP injection in Balb/c mice. The renal sections of 24~48 hours of CP exposure executed no fibrosis and did not grade as it was not significant (≤15%) and found in a patch in some mouse in the corticomedullary regions post 72~120 hours. Glomerulosclerosis was not observed in any of the sections in all groups after CP exposure. The autopsy of dead mice after 120 hours of CP infusion evidenced complete grade 5 (≥75~100% fibrosis) (image not shown) collagen deposition. Hence, mild acute tubular necrosis (ATN) was observed after 24 hours, and severe degenerations were observed after 72 hours of CP infusion. Nearly Grade 4 resulted in extreme damage, and it covered 50% of the damaged area of the corticomedullary region. These parenchymal changes were not observed in mAd-MSCs transplanted mice. mAd-MSCs infusion demonstrated the clear, intact structure of tubular epithelial cells. The damage was attenuated, which was observed with scarce protein casts formed and low inflammatory infiltrates post seven days (***p≤0.001) as compared to controls. Although after mAd-MSCs transplantation, histopathology did not show complete regeneration of all renal tubular cells, and renal tubular dilatation was not significantly diminished, but with a lesser extent of damage survival of mice was increased as compared to CP-treated mice. Some whitish fatty or unfilled empty areas were observed around some tubules, which gave a smooth appearance, and it looked like something had been cleared off in mAd-MSCs treated mice (Fig. 3A, B). The brush border protein loss in AKI was condensed as it was observed with the sub-normal re-expression of Megalin post mAd-MSCs transplantation. Megalin is a specific protein in the brush border of proximal tubular cells. Kim-1 protein expression was reduced after cellular therapy, which is known to express in development and an injury in the kidney. Both expressions were tested by histopathologists in a blind study. It demonstrated that mAd-MSCs infusion protected the kidney to an extent from further severe AKI damage, and these cells can protect from the complete failure of the organ (Fig. 3C).

Fig. 3
mAd-MSCs transplantation restores the kidney architecture. (A) Histopathological improvement in AKI model of balb/c mice in response to cellular therapy by mAd-MSCs. The model was developed by CP nephrotoxicity (18 mg/kg b.w. s.c.). Time course representative kidney sections were stained with Hematoxylin & Eosin (H&E) (raw1), Periodic Acid Schiff’s (PAS) (raw2), and Masson’s trichrome staining (TRI) (raw3), and examined by light microscope. Control (saline) showed the normal architecture of tubules and glomeruli. All changes are shown by arrows. Thick arrows show tubular protein casts formation; thin arrows show degeneration of tubules while double arrows show dilatation of tubules. Congestion (erythrocyte trapping) was displayed in the H&E stain of 24~48 hrs. Some rectangular features of tubules were also observed after 72~120 hrs of CP infusion. Shrinkage of glomeruli was observed in 96 hrs of CP infusion. Both are shown in H&E and PAS stains respectively. Accumulation of nuclei is shown in H&E and PAS stains of 72 hr. PAS stains displayed some sign of fibrosis post 72~120 hrs. All stains exhibited structural improvement post mAd-MSCs transplantation, as well as some clear areas (PAS), were also observed. a=control (saline); b=24 hr; c=48 hr; d=72 hr; e=96 hr; f=120 hr; g=CP+mAd-MSCs. CP: cisplatin, G: Glomerulus, PT: Proximal tubules, DT: Distal tubules (40× Magnification). (B) Representatives graphs of histopathological scoring through H&E and PAS stains for the presence of (a) Tubular epithelial cells’s necrosis or degeneration, (b) Tubular protein casts formation, (c) Infiltration of inflammatory cells, and (d) Renal tubular dilatation. The graphical results are presented as mean±s.d. ANOVA followed by Tucky-Kramer test. *p<0.05, **p<0.01 and ***p<0.001 were the levels of significance for all groups. [1=24 hr; 2=48 hr; 3=72 hr; 4=96 hr; 5=120 hr; 6=CP+mAd-MSCs transplanted]. (C) Immuno changes in Megalin (raw1) and Kim-1 (raw2) expressions, which were enhanced and reduced respectively in mAd-MSCs transplanted mice. These changes exposed the recovery of the renal tubular brush border and reduced necrosis in the renal tissues post cellular therapy. Protein expressions in these sections are represented by arrows. (a) Control (saline); (b) 72 hr; (c) 96 hr; (d) CP+mAd-MSCs transplant (40× Magnification).
![]()
Amelioration of renal dysfunction and improved survival by mAd-MSCs transplantation in CP-induced AKI
Improved survival of AKI balb/c mice attributed to the recovery of renal functions. CP infusion caused a marked increase in serum creatinine, urea, and total urinary protein concomitant to the aforementioned histopathological events. These parameters confirmed the successful development of severely injured renal parenchymal cells in balb/c mice. Significant attenuated (**p≤0.001) mean levels of serum creatinine from <3 fold to~1.5 fold were observed in CP-induced AKI and after mAd-MSCs infusion at day 12 respectively (Fig. 4A). Blood urea and total urinary protein, although did not reach to the significance levels, but they achieved 50% reduction in these levels in post mAd-MSCs transplanted balb/c mice in contrast to CP treatment groups at 96 and 120 hours (Fig. 4B, C). CP toxicity results in 4~8 gram reduction in body weight of Balb/c mice. Most mice died between 96~120 hours, and the percent survival was decreased 80~90% after 96 hours in 18 mg/kg CP-treated mice. Mortality was significantly driven down in mice received mAd-MSCs. Survival of balb/c mice increased, which was observed untill day12 after transplantation of mAd-MSCs and it exposed the efficiency of cell therapy, which is associated with recovery of renal structure and its function (Fig. 4D, E).
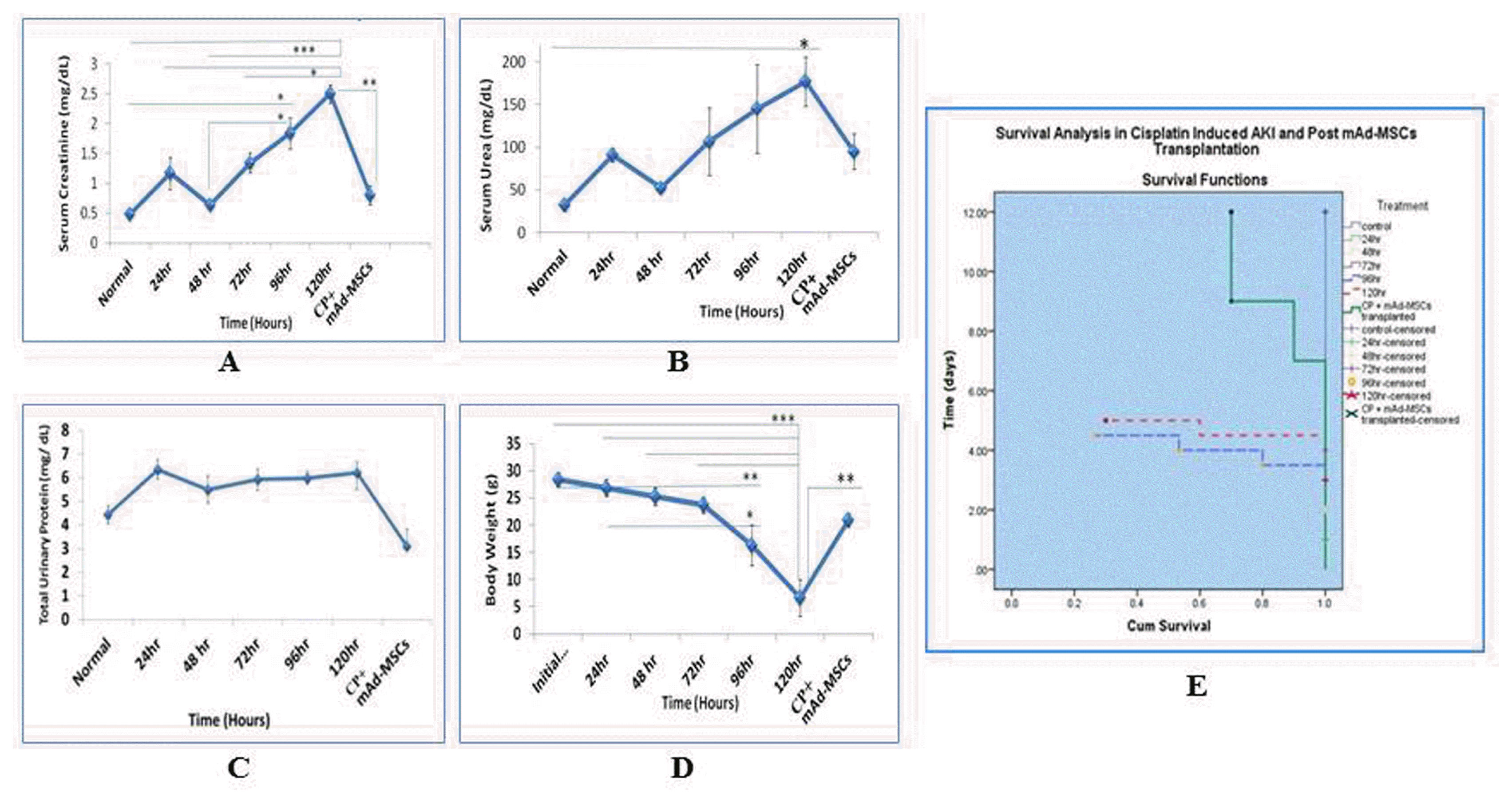
Fig. 4
mAd-MSCs ameliorate kidney failure by restoring its functions, which improved the survival of mice. Functional improvement of kidneys with the survival of balb/c mice was observed in AKI model by CP (18 mg/kg, s.c.) in post transplanted mAd-MSCs. (A) Serum Creatinine, (B) Serum Urea, (C) Total Urinary Protein, (D) Weight dynamics, (E) Survival function for a cumulative survival rate of balb/c mice in all groups was recorded from day 0~12. Data is presented as mean±s.e.m. ANOVA followed by Post-Hoc Tucky’s test for comparison was used. *p<0.5, **p<0.1, and ***p<0.001 were the levels of significance for all groups.
![]()
Regulation of genes in AKI and post mAd-MSCs infused renal tissues
Because MSCs are known to secrete growth factors and have immunomodulatory properties. We screened kidneys by TaqMan real-time quantitative RT-PCR. The changes in the expressions of inflammatory mediator, Tumor necrosis factor alpha (TNF-α); developmental protein, Paired box 2 (Pax2); pro-fibrotic cytokine, Transforming growth factor beta 1 (TGF-β1); survival protein, Mitogen-activated protein kinase 1 (MAPK1); Notch signaling ligand, Jagged1 (Jag1); epithelial markers, Solute carrier family 4 member 2 (SLC4a2); and Kidney specific protein, Ksp-cadherin (Ksp) were evaluated pre and post mAd-MSCs infusion. Inflammatory kidneys of CP-treated animals post 48 hours showed ~7 fold induction of the proinflammatory cytokine; TNF-α. Post mAd-MSCs transplantation demonstrated a marked reduction in TNF-α as compared to pretreated CP infusion. Many studies presented that fibrogenesis was shrunk by decreasing TGF-β 1. TGF-β1 promotes negative regulation in the expression of Pax2 protein through the post-transcriptional mechanism in which it abolished the stability of Pax2 mRNA and consequently, its protein. During embryogenesis, Pax2 is involved in mesenchymal to epithelial transition (MET), i.e. is tubular epithelial cells differentiation, but only confined to collecting ducts in healthy adult kidneys. Pax2 is regulated by TGF-β1. It plays role in tubular cells regeneration, proliferation, tubulogenesis, and apoptosis. Pax2 is re-expressed in proximal tubular cells after injury. In our study peak levels of Pax2 (1×108 fold) significantly up-regulated (***p≤0.001) in 96 hours and when the cells were injected its levels were reduced significantly (***p≤0.001). During CP injury TGF-β1 level was also optimal in 96 hours and exhibited competitive expression vs Pax2. mAd-MSCs presence down-regulated TGF-β1 as well as Pax2 levels (10). Overall MAPK1 advanced to near 40 fold as compared to control, and it attained a normal level post mAd-MSCs infusion. Jagged1 displayed 8 fold changes with respect to control mice. However, there is a very low fold change in Jagged1 of the renal tissues after mAd-MSCs transplantation. Grossly some fold change differences were executed in the expressions levels of SLC4a2 and Ksp. It might be due to the other epithelial cells, which were not affected by CP infusion and were greater than the number of cells in proximal tubules. Hence, these results revealed potential mechanisms whereby administered mAd-MSC exerted their protective effects, which led to significant and prompt renoprotection as a first step (Fig. 5).
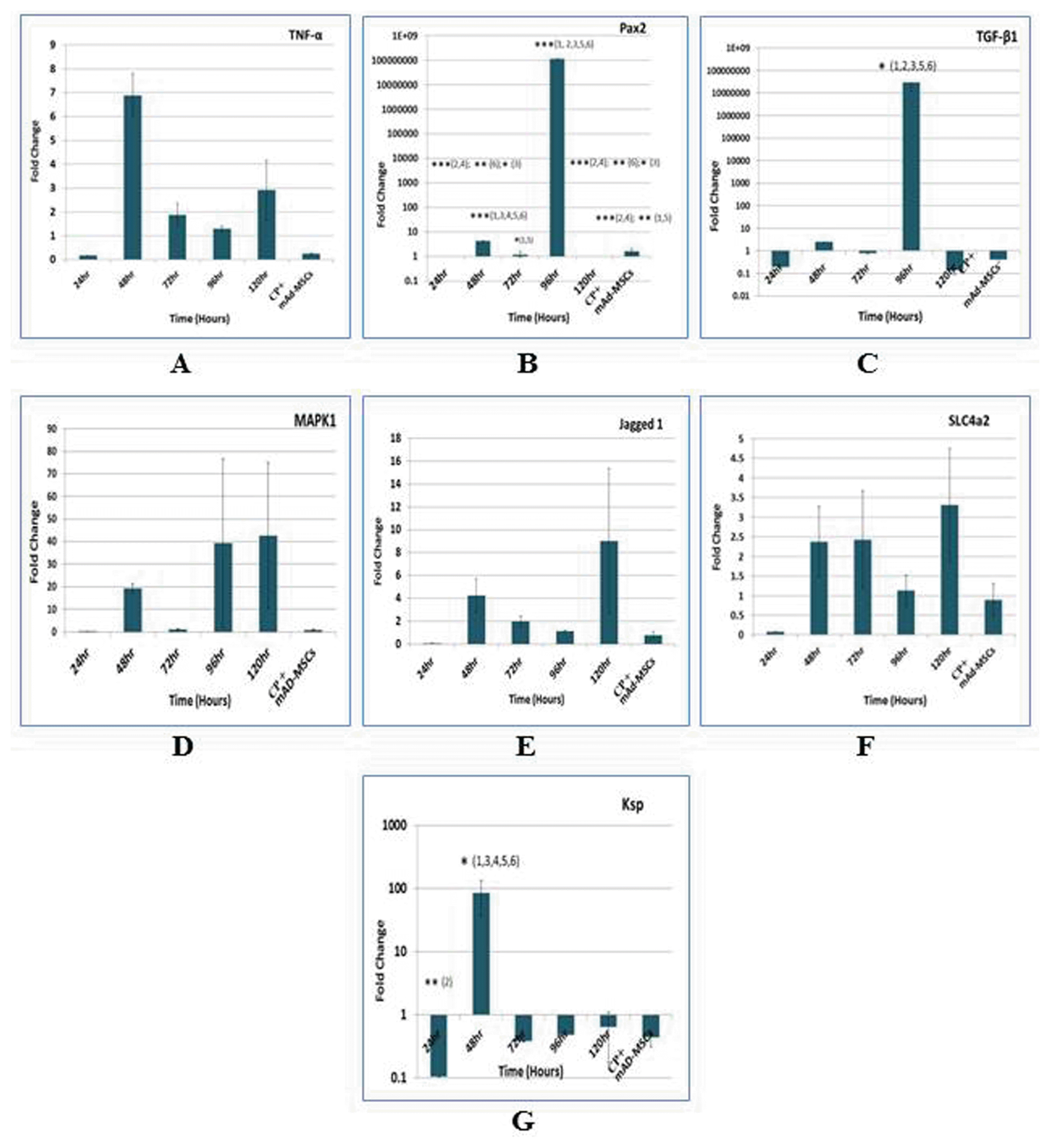
Fig. 5
Transcriptomic features demonstrate the anti-fibrotic and anti-inflammatory effects of mAd-MSCs. Quantitative transcriptomic expressions profiling of the genes in renal tissues of sacrificed balb/c mice by TaqMan qRT-PCR was evaluated in pre (24-120 hours) and post-transplantation of mAd-MSCs. The data was evaluated by the 2^−ΔΔCt method for fold change in genes expressions, which were used for multiple comparisons in ANOVA followed by Tucky’s test. The levels of significance *p<0.05, **p<0.01, and ***p<0.001 were used [log10 scale for Pax2, TGFβ-1, and Ksp]. (A) Tumor necrosis factor alpha (TNF-α). (B) Paired box gene 2 (Pax2). (C) Transforming growth factor beta 1 (TGFβ1). (D) Mitogen-activated protein kinase 1 (MAPK1). (E) Jagged1. (F) Solute carrier family 4 member 2 (SLC4a2). (G) Kidney-specific protein (Ksp-cadherin). [1=24 hr; 2=48 hr; 3=72 hr; 4=96 hr; 5=120 hr; 6=CP+mAd-MSCs transplanted].
![]()
Discussion
AKI is a life-threatening clinical condition, which is a common consequence of ischemic or toxic insults and may lead to complete renal failure or end-stage renal disease (ESRD) (4). mAd-MSCs have multilineage differentiation potential as was demonstrated by osteocytes and adipocytes lineage differentiation. mAd-MSCs were successfully transplanted systemically in Balb/c mice pre-developed with severe AKI through CP. In vivo, CMFDA and Dil fluorescent labeled cells were clearly observed in the kidneys and other organs, but initially within 24~72 hours entrapped in pulmonary capillaries as lungs are the main organs of the vascular system. The entrapment was made temporary, and it was diminished by optimization of cells number, the use of Eskinase (antithrombin), heparin (anticoagulant), and sodium bicarbonate with a very slow rate of cells infusion (2). Through these strategies, pulmonary embolism and thrombosis were not observed. It presented as a safe delivery of mAd-MSCs and 0.5~1×106 mAd-MSCs per mouse was found as a well-tolerated single dose. The liver also demonstrated mAd-MSCs localization as small injuries of CP were also observed in the liver in severe renal failure but no to very little in the heart. Fusion or engraftment of mAd-MSCs was seen within 24~72 hours, predominantly in injured areas of the kidney. mAd-MSCs localization may be engrafted or differentiated into proximal tubular cells for functional restoration of renal parenchyma in vivo. The release of various paracrine factors by mAd-MSCs contributed to tubular cell proliferation. Increase DNA synthesis via BrdU incorporation into replicated DNA in dividing cells indicated that mAd-MSCs promote tubular cell proliferation. It showed the activation of regeneration within a week, and the cells are able to repair the kidneys with CP toxicity.
Nephrotoxicity produced by CP was evidenced morphologically and functionally. CP filtered in glomeruli and secreted in PCT, predominantly in the S3 segment and its uptake via organic cation receptor 1/2 (Oct 1/2). It conjugates with molecules like RNA, DNA, and proteins. CP-DNA binary complex halts DNA synthesis and replication via inter and intrastrand binding to DNA. It results in cell cycle arrest and destruction of intracellular molecules. Reactive oxygen species (ROS) were produced, which collapsed cellular oxidant defense system and cell death occurred (11). PCT is highly affected by CP as it exhibits the highest densities of mitochondria as compared to other nephron’s segments, the main organelle associated with apoptosis. CP triggers apoptosis and necrosis dose-dependently (12). Lower doses 8 μmol/L of CP induces apoptosis while necrotic cell death appeared in few hours of a high dose of 800 μmol/L. In agreement to this in our study a high dose, 1,591.2 μmol/L (18 mg/kg b.wt.) produced severe necrosis in CP-treated mice (13). Consequently, the mononuclear cell infiltrates were observed among renal tubules in compliance to similar studies, which supported that CP nephrotoxicity is mediated by infiltration of inflammatory cells such as neutrophils, macrophages, and the leukocytes as a result of cell death. These cells release pro-inflammatory chemokines and cytokines such as TNF-α and IL-1β. Pro-fibrinogenic factors like TNF-α and TGF-β1 were produced by macrophages, mediates induction of myofibroblastic cells and contributes to the development of fibrosis that is due to excessive production of matrix proteins. Cells organized in spatial patterns demonstrate the underlying functional state. Morphometric examination of kidney sections revealed patchy tubular necrosis, which is associated with tubular brush border or epithelial loss via vacuolization and detachment from the basement membrane, tubular protein casts formation, tubular dilatation, and mononuclear cellular infiltrations. H&E staining signified increased congestion in AKI. Signs of necrosis, i.e. karyorrhexis, karyolysis, and ghost cells were observed. Pyknotic changes indicating apoptosis and necrosis. Glomerular change was started at 96 hours post CP infusion in our study. Proteins like albumin, various enzymes, hormones, and vitamins uptake in proximal tubules are mediated by Megalin (14, 15). Kim-1 is a transmembranous glycoprotein and expressed during development and after ischemic or toxic insult on the surface of proximal tubular epithelial cells. Up-regulation of Kim-1 is considered as a diagnostic marker for ATN and certain types of cancers (16–18). In our study, all the above changes resulted in tubular obstruction, loss of brush border protein; Megalin and induction of Kim-1.
mAd-MSCs transplantation in CP-treated mice displayed renal tissue repair properties by the structural and functional recovery of mice within a week. Corticomedullary regions were recaptured in AKI mice when treated with mAd-MSCs. Further epithelial degeneration, casts formation, and inflammatory exudates were significantly reduced in these renal tissues. The regained tubular structure validated regenerative changes and disappearance of nephrotoxicities like intact cell’s luminal surface, intracytoplasmic vacuoles, and loss of microvilli. Hence, it preserved the tubular epithelium and prolonged the survival of mice. mAd-MSCs produced anti-inflammatory action by repressing cues for TNF-α and lessen hyaline cast (19). Transcriptional expression of TNF-α also denoted these effects. Moreover, Megalin re-expression and reduced expression of Kim-1 confirmed a re-functional state of brush border cells in mAd-MSCs transplanted mice.
Favorable effects of mAd-MSCs as cell therapy were demonstrated in renal functions. Serum creatinine levels attenuated significantly; urea and protein levels were also decreased, which were observed in other models of Ad-MSCs transplantations (20). Therefore, it demonstrated recovery towards kidney functions via restoring kidney architecture, which was discussed in the above paragraph. Further, the body weights of AKI mice, which were slashed 1~2 grams daily but gradually recouped within seven days of mAd-MSCs infusion. These findings indicated that seven days are enough to attain subnormal biochemical levels post infusion of Ad-MSCs.
This study verified an induction of the severe renal injury after CP injection as well as repair and survival of animals after allogeneic mAd-MSCs transplantation. mAd-MSCs administration exerted their effects in AKI by homing or engraftment in renal parenchyma, condensed histological abnormalities, maintenance of the renal structure; consequently, the proliferation of renal cells, post injured functional recovery, and survival of balb/c mice. The mechanisms underlying CP-induced nephrotoxicity and mAd-MSCs transplantation were not evaluated before. Here we are also providing the mechanism through which they produced these effects. A report of 5×105 Luciferase+ -mAd-MSCs transplantation revealed that Luciferase+ -specific mRNA could be detected in injured renal tissue. The induction was significant to 2.2-fold and 6.1-fold higher for the 8 mg/kg, CP and 12 mg/kg CP-induced groups respectively and proved that real-time PCR is a better tool than in vivo bioluminescence imaging (BLI) (21). The change in the expressions of various mediators participates in inflammatory responses, repair, and pathogenesis of AKI was studied in our experimental model of CP-induced AKI alone and after transplantation of mAd-MSCs. The kidney undergoing ATN presents an extensive and complex inflammatory and oxidative stress response caused by toxic insults. It eventually provokes fibroblast proliferation and excessive deposition of the extracellular matrix, that has been recognized as a major contributor to end-stage kidney disease (ESRD). TNF-α is a pleiotropic, pro-inflammatory peptide cytokine that is produced by macrophages, T-cells, and some non-hematopoietic cells. It is cytotoxic in picogram (pg) quantities and promotes marked renal tubular cells apoptosis in response to CP. After DNA platination the first mechanism of DNA damage is the G1 phase cell cycle arrest in renal cells (22). TGF-β1 inhibits BMP7 via down-regulation of E-cadherin. BMP-7 is an important regulator of the tubular epithelial cell function, inflammatory response, and epithelial-mesenchymal transition (EMT). Reduced expression of BMP-7 may contribute to the fibrotic response, and its administration may ameliorate renal injury (1). Consistent with these results, the transplanted cells demonstrated an anti-inflammatory effect by decreasing the expressions of pro-inflammatory cytokines, TNF-α and TGF-β1. There are several studies suggested that MAPK/ERK plays an essential role in AKI (23–26). CP-induced ROS increases the expression of proinflammatory mediators; TNF-α and the activation of MAPK lead to apoptosis in CP-treated rats. We demonstrated the transplantation of mAd-MSCs provides marked protection against CP-induced acute renal damage by negative regulation of MAPK1/ERK2 expression (26). Similar effects on TNF-α and TGF-β1 were observed in the ischemia-reperfusion rat’s model upon 2×106 rat’s Ad-MSCs intravenous cells infusion (20). Anti-inflammatory and anti-fibrotic effects via decreased TNF-α and TGF-β1 culminated in the survival of the mice as compared to CP-treated mice.
Pax-2 has been identified among initial regulators during embryonic development of mammalian kidney from the pronephros through the mesonephros to the metanephros. Expression of Pax-2 in developing kidney induces MET, required for nephric lineage specification. During our studies, there was a gradual increase in Pax-2 expression in adult mice kidneys after CP-induced AKI. The possible reason for the re-expression of Pax-2 on mature tubular epithelial cells during renal injury is hypothesized to take part in the repair of damaged kidney cells, also evidenced by various studies (27–29).
In continuation for demonstrating gene activity in AKI before and after transplantation of mAd-MSCs, the Jagged-1 expression was dramatically increased, which is involved in the developmental pattern or differentiation state of cells. Expressions of Notch receptors and the ligands are decreased in the adult kidney as compared to developing nephrons. Several reports demonstrated that enhanced expressions of Notch receptors and the ligands might contribute to either regeneration or pathogenesis both in acute and chronic kidney injury (30–33). The continuous expression of Notch failed to regenerate complete renal damage, and it is associated with tubule-interstitial fibrosis and glomerulosclerosis. Nyhan et al., 2010 (32) have also shown that TGF-β1 triggered the signaling pathways that regulate Jagged/Notch and EMT-associated genes expressions in human renal tubular epithelial cells and contribute in the development of interstitial fibrosis and glomerulosclerosis. In our studies, the expressions of both TGF-β1 and Jagged-1 were decreased after transplantation of mAd-MSCs, which may delay the progression of end-stage fibrotic kidney disease.
We have also demonstrated for the first time the role of SLC4a2 (anion exchanger) in the pathophysiology of acute renal failure in CP-treated mice. SLC4a2 belongs to the gene subfamily, which encodes Na+ independent anion (Cl−/HCO3−) exchanger proteins (AE2) in mammals (34–37). The mouse AE2 anion exchanger regulates intracellular pH and cell volume and transepithelial ion transport across epithelial cells of many tissues, including renal tubular cells. Acidosis associated with AKI due to the renal damage, predominantly in the tubular cells induces to generate more NH4+, which consequently overexpressed SLC4a2 in CP-treated rats as it is activated by NH4+ and inhibited by protons (38, 39). Besides SLC4a2, we have also noted some change in the expression of a cellular adhesion molecule, Ksp-cadherin. These results are coordinated with the results of Jiang et al., 2004 (40) who evidenced that its expression is disrupted in HgCl2-induced nephrotoxicity as well as in renal failure patients. It is hypothesized that transplantation of mAd-MSCs in CP-treated mice may rescue the tubular damage by inhibiting cellular apoptosis and rebuilding renal architecture, which functionally contributed to maintain the normal acid-base balance as convinced by decreased fold change in the expression of SLC4a2 encoded transporter proteins and Ksp-cadherin. These results were also verified by the improvement in the kidney functions as evidenced by the reduced creatinine and urea levels in CP-treated mice after transplantation of mAd-MSCs.
Finally, the present study demonstrated that the transplantation of mAd-MSCs can protect the renal injury in an experimental CP-induced mice model. These cells function in both differentiation-dependent and independent manners by directly forming renal tubules and exerted pro-proliferative, anti-inflammatory, and anti-fibrotic effects. Our data suggested that mAd-MSCs are a potential cellular source for stem cell-based regenerative therapy for CP-induced AKI.
Conclusions
Conclusively, allogeneic in vitro cultured MSCs infusion is a promising approach to treat AKI. Intravenous mAd-MSCs transplantation displayed the homing potential to the injury site, protective structure, and markedly recaptured renal functions in injured kidneys, which resulted in the survival of mice. The key mechanisms underlying the positive therapeutic impact of mAd-MSC treatment in renal function and pro-proliferation could be due to the localization and suppression of inflammatory and fibrotic cytokine, i.e. TNF-α and TGF-β1 respectively. This study also presented as an approach to get a higher and readily available supply of stem cells. Ad-MSCs transplantation without chemical or genetic manipulation can provide the evidence of therapeutic strategy for the origin of regeneration and overall an improved survival of the system in functionally deprived failed kidneys.
 XML Download
XML Download