

 PDF
PDF Citation
Citation Print
Print


Introduction
Diabetes mellitus (DM) is a group of metabolic diseases characterized by hyperglycemia and excessive urination resulting from insulin resistance or lack of insulin (1). The chronic hyperglycemic condition induces the secretion of advanced glycation end-products (AGEs) from circulating neutrophils, which increases myelopoiesis through the activation of AGE receptor on macrophages and common myeloid progenitors in the bone marrow (BM)(2). As a result, inflammatory monocytes and macrophages increase in the BM infiltrate and migrate into target organs. This induces abnormal functions and structures in micro-vessels, which leads to diabetic vascular complications such as neuropathy, retinopathy, and nephropathy. Thus, circulating and BM hematopoietic stem cells (HSCs) are not only target organs, but are also considered as a contributor to chronic diabetic complications.
In recent years, multiple lines of evidence suggest that diabetes compromises the structure and function of the BM microenvironment. A reduced number of circulating endothelial progenitor cells and HSCs were observed in diabetic human samples (3–5). In addition, diabetic patients showed poor mobilization in response to granulocyte colony-stimulating factor (5, 6). Similar findings have been reported in streptozotocin (STZ)-induced diabetic mice, which is mediated by decreased release of a chemoattractant molecule as well as decreased activation of endothelial nitric oxide (NO) synthase in the BM (7). Kojima et al. (8) reported BM dysfunction including increased fat deposition, reduced hematopoietic tissues, and microvascular rarefaction in diabetic individuals. Ferraro et al. (5) further observed downregulation of C-X-C Motif Chemokine Ligand 12 (CXCL12) in nestin-positive perivascular cells (PVCs). The changes in the BM microenvironment in diabetes are accompanied by functional alterations of hematopoietic cells in the BM. For example, hyperglycemia impaired the colony-forming capability of the BM (9). Downregulation of CXCL12 in perivascular niche cells resulted in the retention of HSCs and their mobilization defects in the BM of diabetic mice (5, 10). Furthermore, in vitro exposure of BM cells to high glucose was found to activate insulin and tumor necrosis factor-α transcripts (11, 12). As mentioned above, it is an emerging hypothesis that diabetes induces hematopoietic alteration and BM niche dysfunction associated with the development of diabetic complications in multiple organs. However, the crosstalk between diabetes and hematopoietic cells has been less investigated.
In our present study, we investigated if hyperglycemic condition influenced hematopoietic composition in the BM and further examined the regenerative potential of PVCs and NADPH oxidase (NOX) inhibition. We found that the hyperglycemic condition induced an increase in the frequency and number of long-term (LT) HSCs as well as the number of total BM cells. The altered hematopoiesis in the BM was partially recovered by the treatment of PVC-derived conditioned medium (CM). Hyperglycemic also increased the number of myeloid-derived suppressor cells (MDSCs) in the BM, which was partially restored by the administration of PVC-CM and NOX inhibitor. We further showed that treatment with PVC-CM and DPI downregulated ERK gene expression and p38 phosphorylation in BM cells of diabetic mice. These findings suggested that the altered hematopoietic composition in the BM by prolonged hyperglycemic conditions might be restored by improving the hematopoietic microenvironment and modulating the activity of NOX.
Materials and Methods
Mice
Mice C57BL/6J mice were purchased from Dooyeol Biotech (Seoul, Korea). Mice were housed in a specific pathogen-free facility. All animal experiments were approved by the Institutional Animal Care and Use Commitment of Kangwon National University. Male C57BL/6J mice (18~20 g, 6 weeks) were intraperitoneally injected with low doses of 50 mg/kg STZ (S0130, Sigma, USA) daily for 5 days to induce type 1 DM (T1DM). After 5 days, blood glucose levels higher than 250 mg/dl were accepted as indicating diabetes induction in the mice. After induction of diabetes, mice were intravenously administered PVC-CM (40 μg/100 μl) for 6 weeks, whereas controls received vehicle.
Preparation of PVC-CM
Human PVCs were seeded in 150 cm2 culture dishes. At 90% confluence, the cells were washed twice with phosphate buffered saline (PBS) and cultured in fresh serum-free α-MEM. After 24 hrs, PVC-CM was collected and filtered through a 0.22 μm filter. Filtered medium were concentrated using a 3-kDa cutoff ultrafiltration membrane and stored at −80°C until use (3K, Amicon).
BM cells harvest and flow cytometry
BM was harvested from femurs by flushing with ice cold RPMI. The cell suspension was centrifuged at 800 g for 5 min, and supernatant aspirated. The cells were resuspended in RBC lysis buffer for 3 min and adding PBS with 1% FBS buffer and once again centrifuged. Cells were incubated with V450 mouse lineage antibody cocktail (561301, BD), in incubation with rat anti mouse Ly-6A/E (Sca-1) (558162, BD) and Rat anti mouse CD117 (c-kit) (553356, BD) antibodies, to quantify the percentage of Lin− Sca1+cKit+ (LSK) Cells. Another population of cells from BM was stained with rat anti mouse CD34 (560238, BD) to identify the population of LT-HSCs and short-term (ST)-HSCs. The antibodies used for the MDSCs experiments were CD11b-PEcy7 (25-0112-82, eBioscience), Ly6G-eflur 450 (45-5931-82, eBioscience), and Ly6C-APC (17-5932-82, eBioscience). All antibodies were incubated for 30 min at 4°C and samples were analyzed with FACS Canto II (BD).
Colony-forming unit (CFU) assay
CFU assay was performed as previously described (13). Briefly, 6,000 BM cells were plated into methylcellulose H4434 (Stem Cell Technologies) and incubated for 7~10 days at 37°C in 5% CO2.
Real time qRT-PCR
Total RNA from lung tissues was extracted using an RNeasy Mini Kit (Qiagen, Duesseldorf, Germany) and the cDNA was synthesized using TOPscriptTM RT DryMIX (Enzynomics, Daejeon, Korea). qPCR analyses were performed using a Step One Plus real time PCR system (Applied Biosystems, Warrington, UK) with TOPrealTM qPCR 2X PreMIX (Enzynomics). To amplify GAPDH, arginase-1 (ARG1) and inducible nitric oxide synthase (iNOS), we used the following primers: GAPDH forward (F) 5′-AACTTTGGCATTGTGGAAGG-3′, reverse (R) 5′-ACACATTGGGGGTAGGAACA-3′, ARG1 F 5′-ATGCAAGAGACCTTCAGCTAC-3′, R 5′-GCTGCTTTCCCAAGAGTTGGG-3, and iNOS F 5′-GGCAGCCTGTGAGACCTTTG-3′, R 5′-TGAAGCGTTTCGGGATCTG-3′.
Western blotting
Lysates from BM total cells were lysed in protein lysis buffer with protease inhibitor (#1860932, Thermo Scientific) and quantified using the BCA protein assay (#23228, Thermo Scientific). The 20 μg of protein were separated by SDS-PAGE using 10~15% gel and then transferred to PVDF membranes (IPVH00010, Millipore, Billerica, MA, USA). Membranes were blocked with 5% skim milk for 1h at room temperature and then incubated with primary antibodies against anti-phospho-p44/42 MAPK (4370s, Cell Signaling), anti-p44/42 MAPK (4695s, Cell Signaling), anti-phospho-p38 MAPK (4511s, Cell Signaling), anti-p38 MAPK (9212s, Cell Signaling), anti-phospho-SAPK/JNK (4668s, Cell Signaling), anti-SAPK/JNK (9252s, Cell Signaling) and their corresponding secondary antibodies (anti-rabbit BML-SA204-0100, anti-mouse ADI-SAB-300-J, Enzo) overnight at 4°C and for 1 h at room temperature, respectively. Membranes were scanned with ChemiDoc Imaging System (Bio-Rad Laboratories, Hercules, CA, USA).
Results
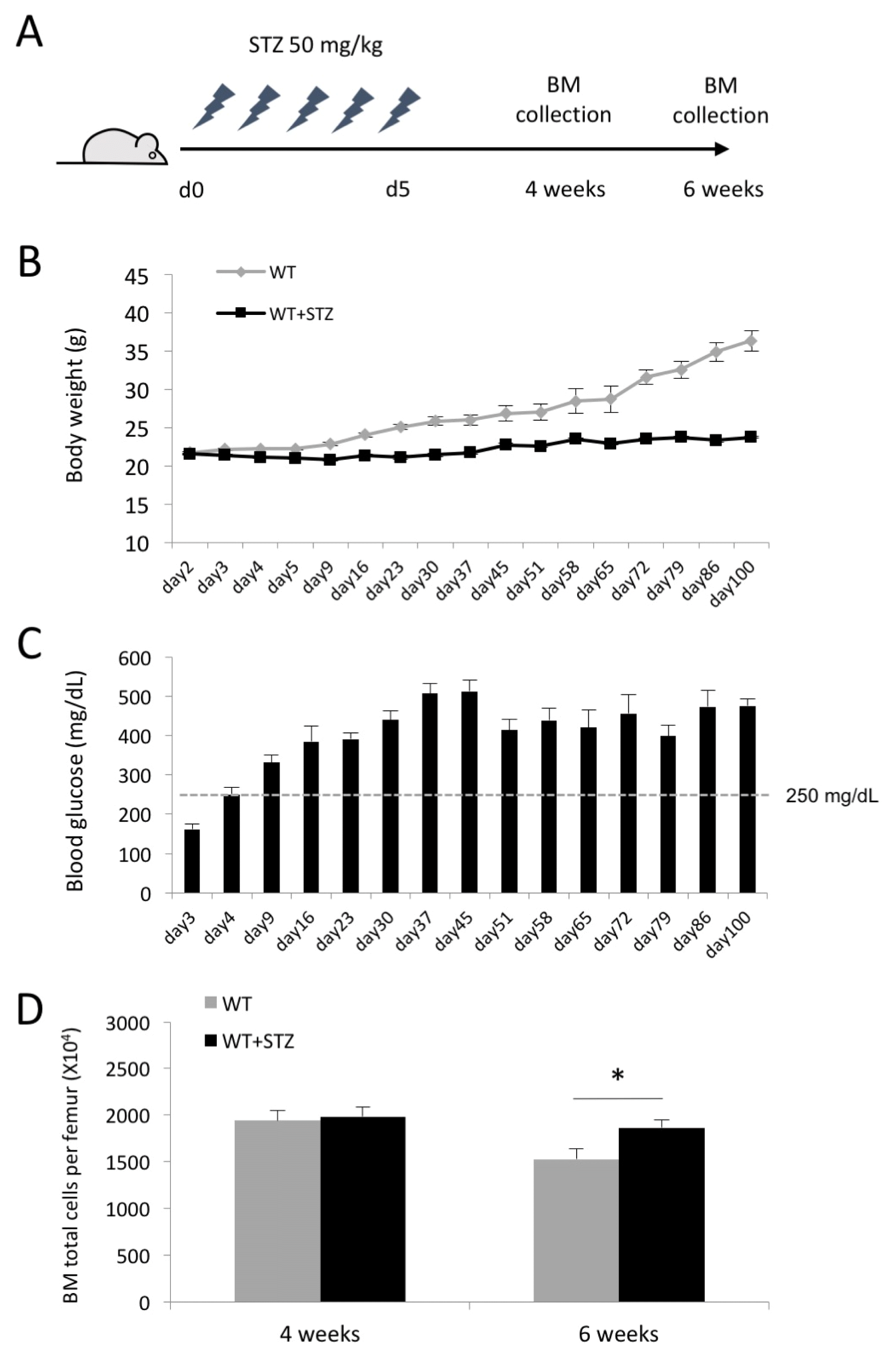
Hyperglycemic conditions increase the number of BM cells
To investigate the effects of hyperglycemic conditions on hematopoietic composition in the BM, type I diabetic mice were induced by daily injection of STZ for 5 days and sacrificed 4 and 6 weeks after the last injection of STZ (Fig. 1A). The STZ-injected mice exhibited gradual weight loss during the time course of experiments compared to the control mice (24.5±1.5 g vs. 28.37±1.6 g at 6 weeks after the onset of diabetes) (Fig. 1B). The blood glucose level in diabetic mice was maintained ≥250 mg/dL up to 100 days after the onset of diabetes (Fig. 1C). We harvested BM-derived mononuclear cells at 4 and 6 weeks after induction of diabetes and first determined the impact of hyperglycemic conditions on the total number of BM cells. The total number of BM cells was significantly increased in diabetic mice compared to the control mice at 6 weeks after induction of diabetes, while there was no difference in the number of BM cells at 4 weeks (Fig. 1D). This finding suggested that prolonged hyperglycemic conditions may alter the hematopoietic composition in the BM.
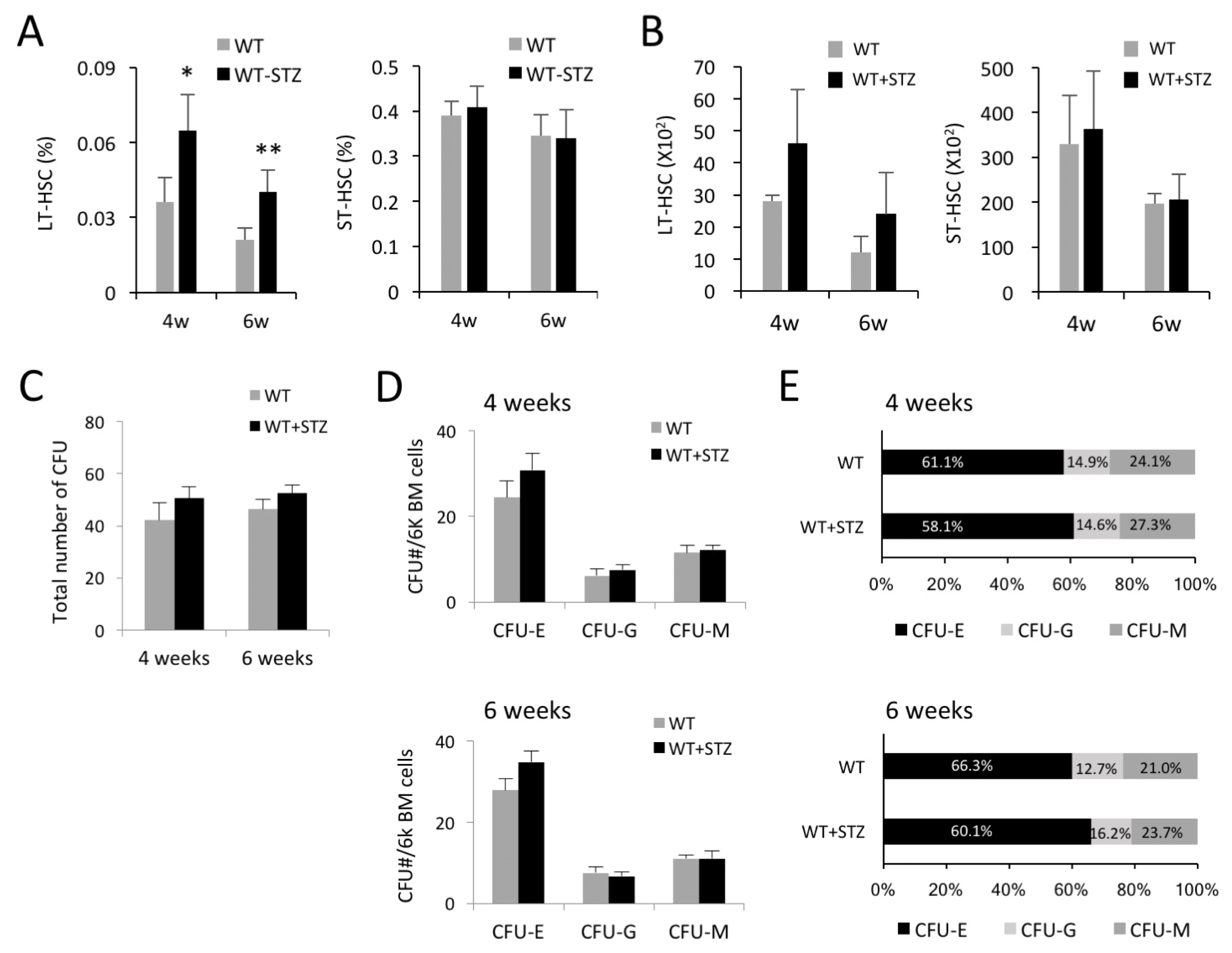
Percentage and number of LT-HSCs are increased in STZ-injected mice
We assumed that an increased number of total BM cells was reflected in the hematopoietic composition in the BM. Therefore, we first investigated the impact of hyperglycemia on the frequency and number of LT- and ST-HSC populations at 4 and 6 weeks after induction of diabetes. The frequency of LT-HSCs (Lin−Sca1+cKit+ CD34−) was not changed at 2 weeks after induction of diabetes (data not shown) but increased at 4 weeks and was retained up to 6 weeks, whereas the frequency of ST-HSCs (Lin−Sca1+cKit+CD34+) was not affected by hyperglycemic conditions (Fig. 2A). We analyzed the number of LT- and ST-HSCs in the BM and similarly found an increase of the LT-HSC fraction at 4 and 6 weeks after induction of diabetes, but no changes in the ST-HSC fraction (Fig. 2B). We then determined the effects of diabetes on the colony-forming ability of BM-derived HSCs. Total numbers of colony-forming units (CFUs) and CFU subtypes were not significantly different between the STZ-injected and the control mice, while the percentage and number of CFU-E showed an increased trend without reaching significance (Fig. 2C–E). These results suggested that hyperglycemia not only affected self-renewal or proliferative activity of LT-HSCs in the BM, but also may have influenced their functional characteristics.
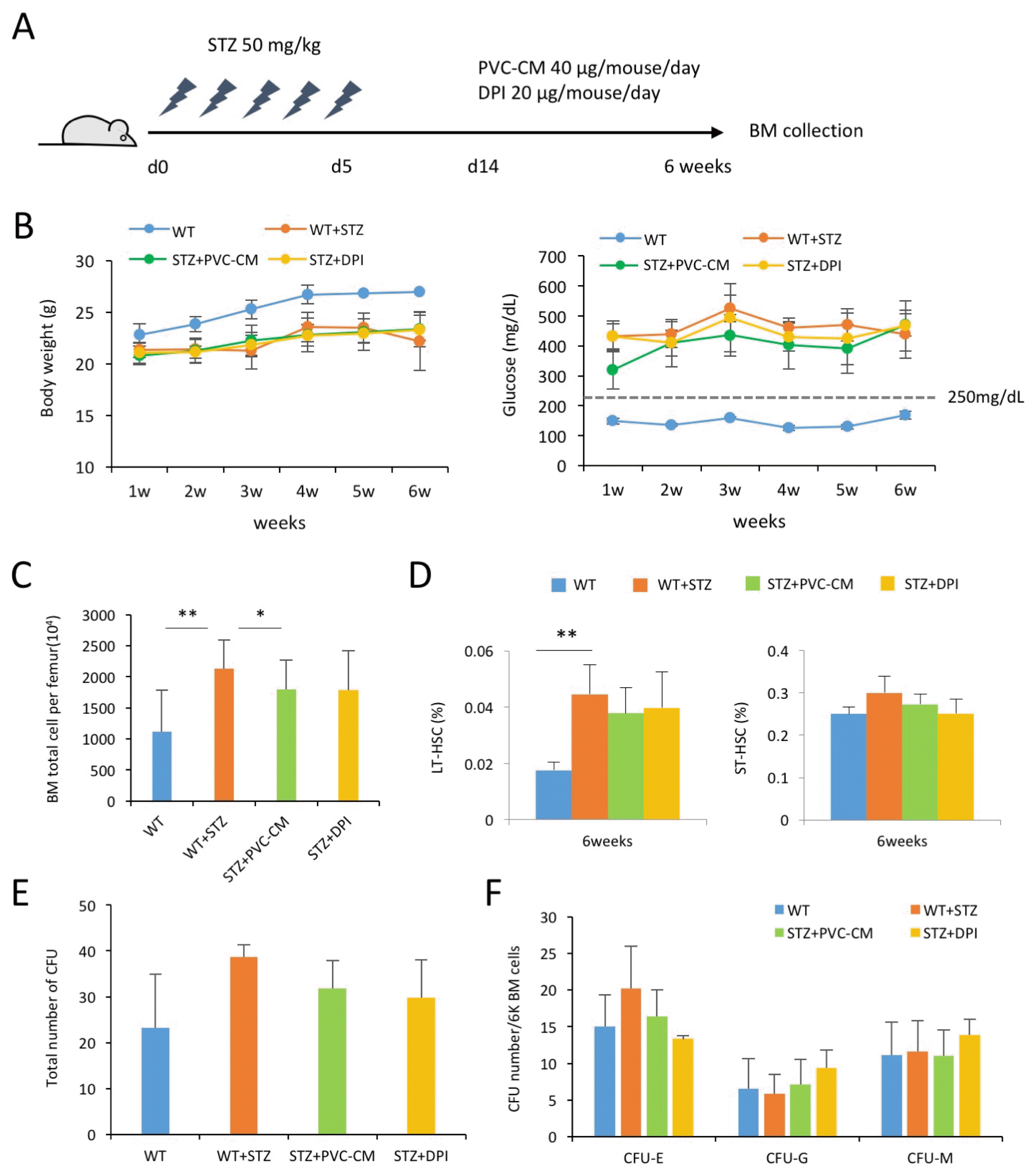
Alterations in the number of total BM cells of diabetic mice partially revert to normal by PVC-CM treatment
Previous studies indicated that diabetes impaired the structure and function of the BM microenvironment, which is mechanically mediated by activation of inflammatory responses and generation of reactive oxygen species (ROS) via a NOX system. Thus, we further determined if perivascular niche support and an inhibition of NOX recovered the hematopoietic alteration induced by hyperglycemic conditions. After induction of diabetes, the mice daily received either 40 μg PVC-CM or 20 μg diphenyleneiodonium (DPI, NOX inhibitor) for up to 6 weeks (Fig. 3A). Administration of PVC-CM or DPI did not recover the loss of weight and reduce the increased blood glucose level in STZ-treated mice (Fig. 3B). The increased number of total BM cells in diabetic mice was partially reverted to normal when PVC-CM was administered (Fig. 3C), whereas both PVC-CM and DPI did not reduce the increased frequency of LT-HSCs (Fig. 3D). Furthermore, an increased trend in the total number of CFUs and CFU-E in STZ-treated mice partially reverted to normal in both PVC-CM and DPI-injected mice (Fig. 3E and 3F). These results indicated that hyperglycemia may have impaired the BM microenvironment and stimulated the NOX system, which could be partially restored by perivascular support and inhibition of NOX activity.
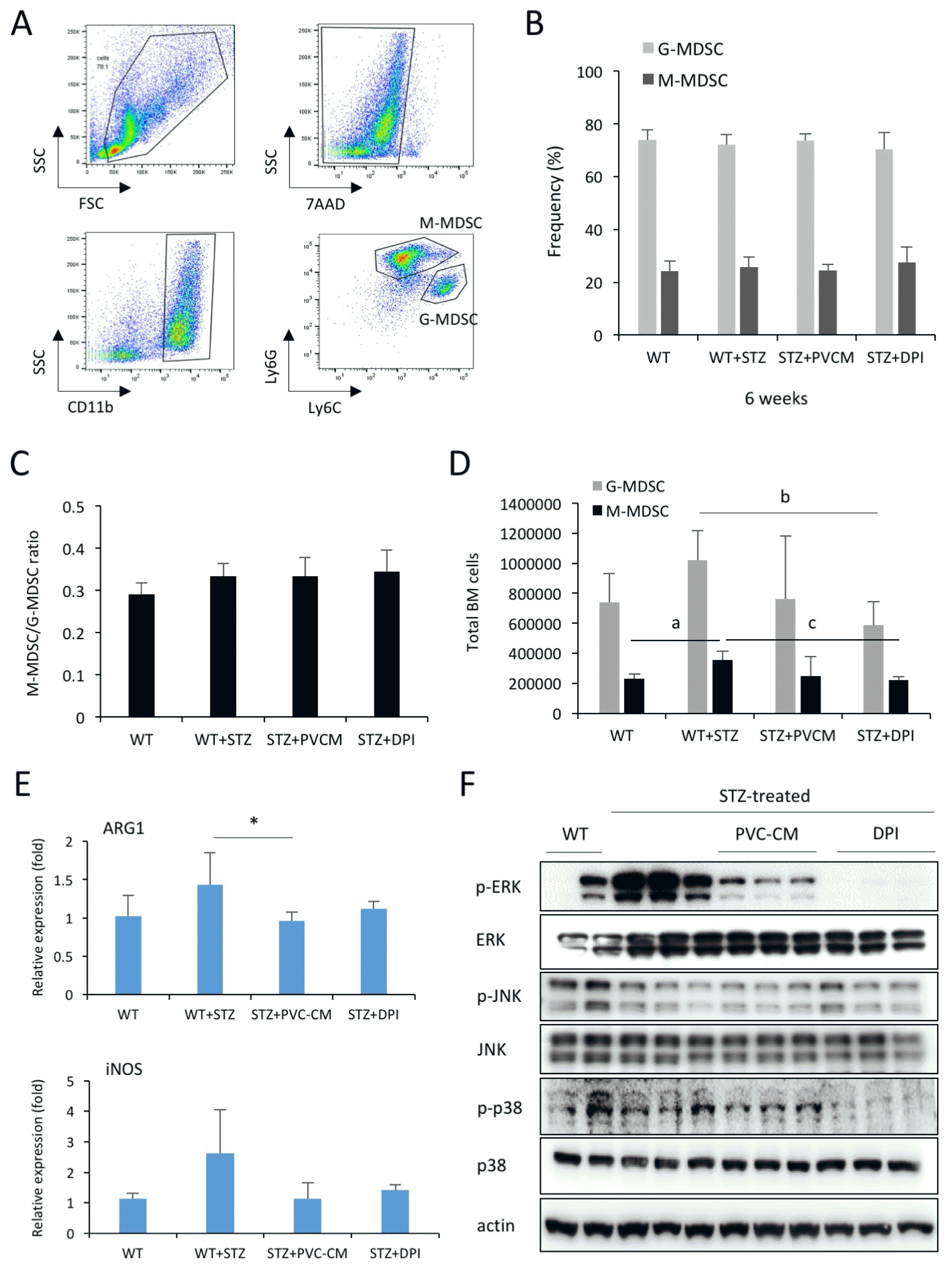
PVC-CM treatment modified the expansion of MDSCs population by hyperglycemia
A recent study reported a key role of T cells in the progression of type 2 DM (T2DM), which can be suppressed by MDSCs (14). Therefore, we next asked if the hyperglycemic condition affected the number and frequency of MDSCs in the BM. Mouse MDSCs were divided into two subtypes based on expression of surface markers as follows: CD11b+Ly6G+Ly6Clow granulocytic MDSCs (GMDSCs) and CD11b+Ly6G−Ly6Chi monocytic MDSCs (M-MDSCs) (Fig. 4A) (15, 16). We found that hyperglycemic conditions did not alter the ratio between G-MDSCs and M-MDSCs as well as the frequencies of G-and M-MDSC subsets (Fig. 4B and 4C). The treatment of PVC-CM and DPI did not influence the frequency and ratio of both cell types. Interestingly, hyperglycemic conditions significantly increased the number of G- and M-MDSCs in the BM, which was efficiently restored by the administration of PVC-CM and DPI (Fig. 4D). ARG1 and iNOS are highly produced by MDSCs (17, 18) and involved in immunosuppressive events in pathologic condition. Thus, we examined whether PCV-CM and DPI treatment would influence the transcript levels of ARG1 and iNOS in MDSCs. PCR data showed that their levels were decreased by PVC-CM and DPI treatment (Fig. 4E). To understand the molecular mechanism of the alterations in hematopoietic composition, we examined the expression levels of MAPKs in the BM cells of the control and diabetic mice. We found the activation of ERK and p38 phosphorylation in STZ-treated mice compared to the control mice, which was completely reversed to normal after PVC-CM and DPI treatment (Fig. 4F). However, expression of JNK was not changed by hyperglycemic conditions and PVC-CM/DPI treatment. These findings suggested that alterations in the number of MDSCs and activation of MAPK signaling molecules might be associated with functions of MDSCs and subsequent diabetic complications.
Discussion
Cellular niches in the BM are functional compartments that control the behavior of HSCs and immune cells via direct and indirect signal transmission. The results of our study showed that prolonged hyperglycemic conditions altered the hematopoietic composition in the BM, which was partially restored by the paracrine support of perivascular niche cells and modulation of NOX activity. This finding suggested that DM influenced the function and differentiation of hematopoietic cells by impairing the BM perivascular niche.
Since the concept of BM niche was first proposed (19), multiple types of niche components and their regulatory factors have been relatively well defined (20). First, the bone-forming osteoblasts secret various hematopoietic cytokines and adhesion molecules that contribute to the maintenance and differentiation of HSCs. Second, vascular cells including endothelial cells and PVCs were found to express soluble factors that promote hematopoiesis and adhesion molecules such as E-selectin and VCAM1 that facilitate cell homing (21). Third, subpopulations of mesenchymal cells including CD146+ adventitial cells, CSCL12-abundant reticular cells, and adipocytes have been identified as BM niche components that regulate HSC function (22, 23). Among these cellular niches, PVCs have been consistently shown to function as a pivotal supporter in HSC maintenance (24, 25), illustrating the crosstalk of the PVCs and HSCs in the BM microenvironment. PVCs are one of the best contributors to maintaining HSCs in the BM, which can support LH-HSCs by binding with Notch ligand Jagged-1 (26, 27) suggesting close relationship with HSCs in reconstituting blood lineage cells. As shown in previous studies, hyperglycemia conditions resulted in the loss of PVCs in several tissues via accumulation of AGEs, high levels of VEGF, and ROS production (28, 29), suggesting relevance to vascular defects in diabetes. In our previous study, PVCs from the patients undergoing gestational diabetes showed significant defects in growth and regeneration potential (30). All these findings clearly indicate that hyperglycemic conditions influence the function and proliferation of HSCs by compromising the BM perivascular niche. Our results showed that the hyperglycemia-induced alteration in hematopoietic composition was partially recovered by the administration of PVC-CM. Thus, identification of specific regulatory soluble factors from PVCs is essential to apply disease-specific preconditioning treatments (31).
MDSCs originate from the myeloid lineage, but can be discriminated from other myeloid cell types by their immunosuppressive capacity. Their suppressive role is exerted through increased production of ARG1, NO and ROS in pathological conditions. We found that MDSC number and expression level of ARG1 and iNOS transcripts in the BM were significantly increased. These changes of MDSCs in the BM may play an important role in preventing or alleviating the progression of diabetic mellitus by suppressing T cell activity. Recent studies support the potential role of MDSCs in DM. For example, MDSCs are rapidly enriched in peripheral blood of both mice and human with T1DM (32, 33). Wang et al. (14) also showed that MDSCs prevent the development of T2DM by suppressing CD4+ T cell activity. These findings suggest that strategies to enhance MDSC suppressive function and number may be effective in preventing or alleviating DM.
Insulin signaling is a highly conserved metabolic process and diabetes accompanying with insulin resistance is caused by downregulation for the insulin receptor gene following persistent MAPK/ERK pathway inhibition (34). In addition, JNK and p38 are known as linked signal factors in diabetes (35, 36). Consistent with previous data, we also showed that phosphorylated ERK and p38 were activated in diabetic BM cells. However, and the levels of ERK and p38 were rapidly decreased by PVC-CM and DPI, implying that soluble factors from PVCs may be relevant to ERK and p38, but not JNK in restoring the microenvironment during diabetes. Further study is required to identify soluble factors from PVCs and define their roles in preventing or treating diabetic complications by reducing hematopoietic alterations in the BM.
 XML Download
XML Download