

 PDF
PDF Citation
Citation Print
Print


Introduction
Imprinted genes, which are regulated by parental-specific epigenetic marks such as DNA methylation, are important in mammalian fetal growth and development (1). Notably, most imprinted genes are found in the brain. Dysregulation of these genes in the brain can lead to developmental disability, cognitive impairment, speech impairment, and behavioral problems (2, 3). Genomic imprinting varies in a tissue- and parent-of-origin-specific manner. Differentially methylated regions (DMRs) in imprinted genes also vary in a tissue-specific manner. Especially, maternal DMRs have more variable methylation levels in somatic tissue than paternal DMRs (4). Differential expression of imprinted genes may occur during development. In mouse, imprinted genes are expressed in different proportions in the fetal brain and adult brain (5). Therefore, the genomic imprinting status in various neural cells developing embryo needs to be examined for understanding gene expression and function of imprinted genes in a tissue or cell type-specific manner.
To understand the function of imprinted genes and the link between these genes and neurogenetic disorders, many studies have used animal models with genetic mutations. However, these models may not accurately recapitulate human genotypes and cellular phenotypes because of the difference in proliferation rates between mouse and human (6). Human uniparental pluripotent stem cells, in which both alleles are inherited from the one parent, are useful for investigation of genomic imprinting and the role of imprinted genes during development (7). Nevertheless, the use of human embryonic stem cells (ESCs) remains an ethical issue in many countries.
In the present study, we describe genomic alterations of imprinted genes during reprogramming and differentiation of neural stem cells (NSCs) derived from human parthenogenetic induced pluripotent stem cells (hPgi-PSCs) that originated from a benign ovarian teratoma (dermoid cysts). Stelzer et al. (8–10) have reported that hPgiPSCs obtained from dermoid cysts are useful for investigation of genomic imprinting. Our previous study identified novel imprinted single CpG sites presenting a parent-of-origin-dependent status using hPgiPSCs and also demonstrated that hPgiPSCs are useful tool to investigate genomic imprinting in humans (11). In this study, we analyzed DNA methylation and gene expression and observed dynamic alterations on maternal alleles that were consistent with published data for in vivo mouse models and patient samples. Moreover, the alteration of genomic imprinting status differentially showed each neural cell types. Therefore, the model established in this study can be used as a human in vitro model to study genomic imprinting and the roles of imprinted genes in neurodevelopment and neurogenetic disorders.
Materials and Methods
Human induced pluripotent stem cells
Human parthenogenetic fibroblasts were obtained from mature cystic ovarian teratoma tissues from elective surgeries with female patient consent as approved by the Konkuk University Medical Center, Seoul, Korea (KUH-1040045) (11). Human somatic fibroblasts were obtained from adipose tissue from elective surgeries with female patient consent as approved by the Institution Review Board of Pusan National University Hospital, Pusan, Korea (H-2008-116) (12). iPSCs were generated as previously described (11). Briefly, somatic and parthenogenetic fibroblasts were transfected using retroviral vectors, OCT4, SOX2, KLF4, and c-MYC. Established iPSCs were cultured on a Matrigel (Corning, NY, USA)-coated plate in mTeSRTM 1 medium (StemCell Technologies, Vancouver, Canada).
Derivation of neural stem cells from human induced pluripotent stem cells
For embryoid body (EB) formation, the iPSC colonies were removed from culture dishes with Accutase (Sigma-Aldrich, MO, USA) and transferred into non-coated 60 mm culture dishes (SPL Life Science, Pocheon, Korea). The EBs were maintained in mTeSR1 with a ROCK inhibitor for 4 days. For rosette formation, the EBs were plated on a T-25 flask coated with 0.2% gelatin in neural induction medium [DMEM/F12 with 1×N2 supplement (Invitrogen, MA, USA), 2×B27 supplement (Invitrogen), 10 ng/ml human epidermal growth factor (Prospec, NJ, USA), and 10 ng/ml human fibroblast growth factor 2 (FGF2; Peprotech, Seoul, Korea)] for 10~14 days. The rosettes were mechanically isolated and transferred into non-coated 12-well plates for 7 days in rosette floating medium [DMEM/F12 with 1×N2 supplement and 10 ng/ml FGF2]. The neurospheres were plated on 6-well plates coated with laminin (5 ng/ml) in expansion medium [DMEM/F12 with 1×N2 supplement, 1×B27 supplement without vitamin A (Invitrogen), 1×Gem 21 supplement (Gemini Bio Products, CA, USA), and 10 ng/ml FGF2]. A typical split ratio was between 1:2 and 1:3 with high density (every 3 days) and the medium was changed every 2 days.
Differentiation of neural stem cells
For neuron differentiation, NSCs were plated on poly-L-ornithine (Sigma-Aldrich)/laminin-coated 4-well plates (1×104 cells/well) in neuron differentiation medium [DMEM/F12: neural basal medium (Gibco, MA, USA), 50:50 with 1×N2 supplement, 1×B27 supplement, and 300 ng/ml of cyclic AMP] for 5 weeks. Astrocyte differentiation was performed in 3 steps. The medium for step 1 was KnockOut DMEM/F12 (Invitrogen) with StemPro Neural Supplement (Invitrogen), 10 ng/ml Activin A (Prospec), 10 ng/ml Heregulin 1β (Prospec), and 200 ng/ml insulin-like growth factor I (Prospec), and was changed daily for 2 weeks. The medium for step 2 was NSC expansion medium with 10 ng/ml bone morphogenetic protein 4 (Prospec) and 8 ng/ml FGF2 for 2 weeks. For step 3 (maturation), the cells were cultured in maturation medium (XCell Science Inc., CA, USA) for 3 weeks.
RT-PCR and quantitative real-time PCR
We used the RNeasy Kit (Qiagen, Hilden, Germany) to extract total RNA following the supplier’s instructions. Total RNA (1 μg) was reverse-transcribed into cDNA using the Omniscript RT Kit (Qiagen) following the manufacturer’s protocol. Total mRNA of human fetal brain (TaKaRa, Shiga, Japan) was used as a positive control to assess the expression of NSC-specific markers in derived NSCs. All RT-PCR mixtures contained Ex Taq Polymerase (TaKaRa); PCR was performed for 30 cycles for all markers. Imprinted gene expression levels were evaluated by quantitative real-time PCR using a 7500 Real-Time PCR System (Applied Biosystems, CA, USA) and SYBR Green (Enzynomics, Daejeon, Korea). All primer sequences are listed in Supplementary Table 1.
Immunocytochemistry
Cells on glass coverslips were fixed in 4% paraformaldehyde for 15 min at room temperature and washed with 1×Dulbecco’s phosphate buffered saline (DPBS; Hyclone Laboratories, UT, USA). Immunocytochemistry was performed as previously described (13). Antibodies and their dilutions are listed in Supplementary Table 2. The nuclei were counterstained with DAPI (Sigma-Aldrich). The cells were analyzed with a fluorescence microscope (Olympus, Tokyo, Japan).
Genomic DNA isolation and bisulfite treatment
Genomic DNA was isolated using a G-spin Total DNA Extraction Kit (iNtRON, Seongnam, Korea). Genomic DNA (1 μg) was modified using an EpiTect Bisulfite Kit (Qiagen) according to the manufacturer’s instructions.
Methylation-specific PCR
To perform methylation-specific PCR, bisulfite-converted DNA was used to seed a multiplex using the primers that confirmed as previous study (14), 15q maternal and 15q paternal, in Supplementary Table 1. Maternal and paternal controls were performed by using maternal or paternal primer sets alone on previously amplified templates. The maternal fragment size is 174-bp, and the paternal fragment size is 100-bp.
DNA methylation analysis using bisulfite sequencing
DNA methylation status was analyzed using bisulfite sequencing analysis as previously described (13). The analyzed region containing imprinted genes is presented in Supplementary Table 3. The data were visualized and aligned using QUMA (Quantification tool for Methylation Analysis; http://quma.cdb.riken.jp/).
Statistical analysis
The values are reported as mean±SEM. All statistical tests were performed using the GraphPad Prism Software version 5.00 for Windows (www.graphpad.com). For each cell type, significant differences between somatic and parthenogenetic groups were evaluated by analysis of variance (ANOVA) followed by a two-tailed t-test. Significance of inter-group differences was analyzed using one-way ANOVA.
Results and Discussion
Derivation and characterization of parthenogenetic neural stem cells
Differentiation of pluripotent stem cells into NSCs is considered an in vitro model to study nervous system development during embryogenesis (15). We differentiated hPgiPSCs into NSCs according to the procedure shown in Fig. 1A to establish a human in vitro model to investigate alterations of genomic imprinting in neurodevelopment (Fig. 1A). NSCs were derived using the EB formation method and the cells were obtained by mechanical dissociation of neural rosettes. To confirm the differentiation potential, we differentiated the hSi-NSC and hPgi-NSCs into neurons and astrocytes under same condition and periods. In both hSi-NSCs and hPgi-NSCs, RT-PCR analysis showed a decreased expression of pluripotency marker genes (OCT4 and NANOG), and expression of self-renewal and NSC marker genes (SOX2, SOX1, PAX6, and MASH1) (Fig. 1B). Using immunocytochemistry, we also confirmed that hSi-NSCs and hPgi-NSCs expressed the NSC marker proteins SOX2 and PAX6 (Fig. 1C). hPgi-NSCs could subsequently differentiate into neurons and astrocytes. Neurons differentiated from hSi-NSCs and hPgi-NSCs expressed two neuronal marker proteins, microtubule-associated protein 2 (MAP2) and class III beta-tubulin (TUJ1), and astrocytes differentiated from hSi-NSCs and hPgi-NSCs expressed a mature astrocyte marker protein, glial fibrillary acidic protein (GFAP) (Fig. 1D). These data show that hPgiPSCs, which have only maternal alleles, can be differentiated into NSCs and further differentiated into neurons and astrocytes, and that the established hPgi-NSCs have normal characteristics similar to those of hSi-NSCs.
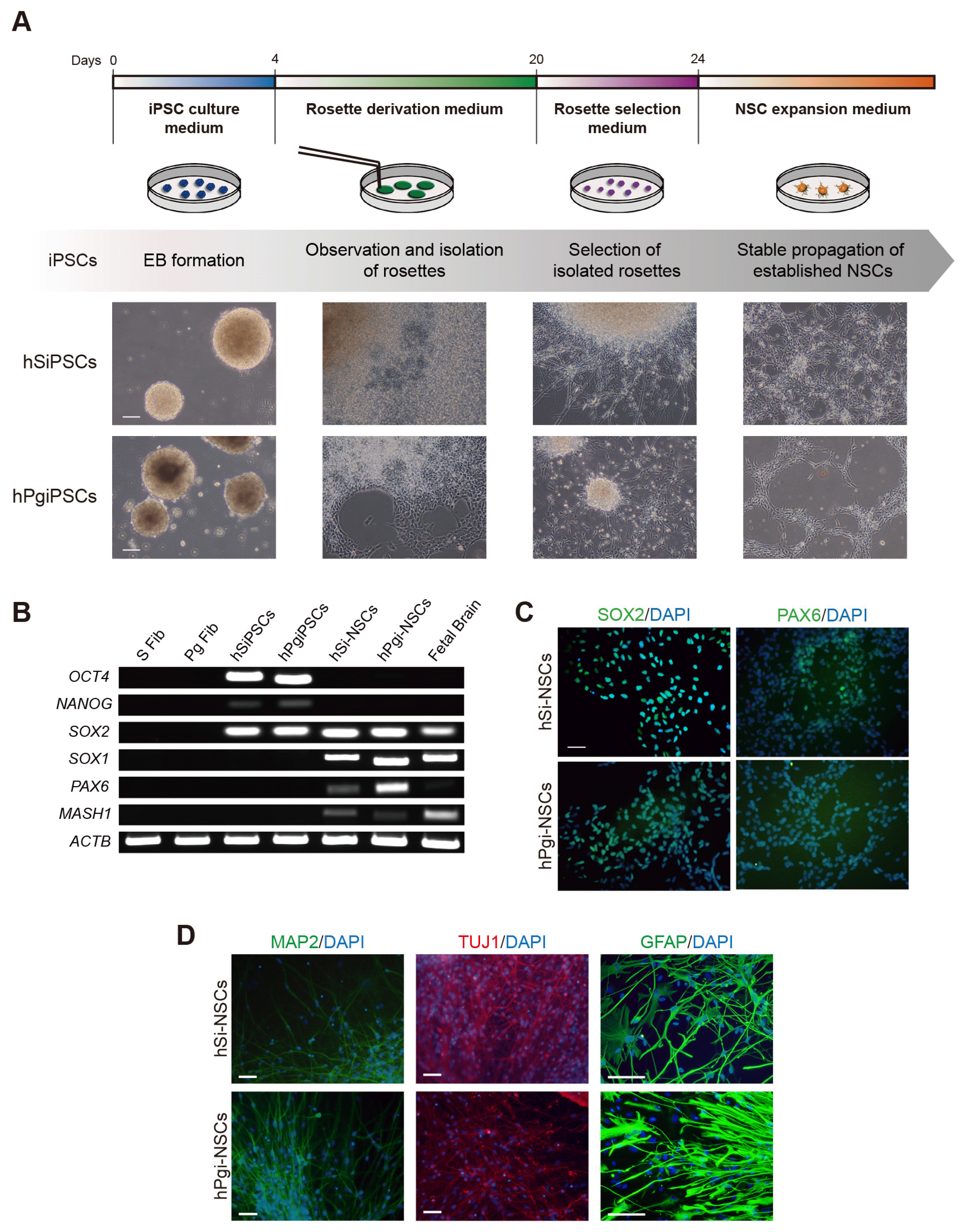
Fig. 1
Derivation of NSCs from iPSCs and their characterization. (A) Schematic representation and phase contrast photomicrographs of hSi-NSCs and hPgi-NSCs derivation. hSi-NSCs, human somatic induced pluripotent stem cell-derived neural stem cells; hPgi-NSCs, human parthenogenetic induced pluripotent stem cell-derived neural stem cells. (B) RT-PCR analysis of the expression of pluripotency markers (OCT4 and NANOG), and self-renewal and NSC-specific markers (SOX2, SOX1, PAX6, and MASH1) in S Fibs, Pg Fibs, hSiPSCs, hPgiPSCs, and in differentiated hSi-NSCs and hPgi-NSCs. S Fibs, human somatic fibroblasts; Pg Fibs, human parthenogenetic fibroblasts; hSiPSCs, human somatic induced pluripotent stem cells; hPgiPSCs, human parthenogenetic induced pluripotent stem cells. (C) Immunocytochemistry of hSi-NSCs and hPgi-NSCs shows the expression of NSC marker proteins (SOX2 and PAX6; green), which co-localize with DAPI (blue) in the nucleus. Scale bar: 100 μm. (D) Immunocytochemistry of neurons and astrocytes differentiated from hSi-NSCs and hPgi-NSCs shows the expression of neuron marker proteins (MAP2, green; TUJ1, red), and an astrocyte marker protein (GFAP, green), which co-localize with DAPI (blue) in the nucleus. Scale bars: 100 μm. MAP2: microtubule-associated protein 2, TUJ1: neuron-specific class III beta-tubulin, GFAP: glial fibrillary acidic protein.
![]()
Dynamic alteration of dNA methylation of imprinted genes during reprogramming and neural differentiation
Several imprinted genes in the brain are important for cell proliferation, differentiation, and normal function of the nervous system. The loss of function of these genes in the brain has severe consequences for behavior in humans (2). Mammalian development requires proper expression of imprinted genes. Expression of imprinted genes are largely regulated by an epigenetic modification such as DNA methylation in CpG islands in a parent-of-origin-specific manner (16). Therefore, it is important to investigate changes of DNA methylation patterns of imprinted genes during neurodevelopment. To do so, we examined DNA methylation during neural differentiation.
We analyzed the DNA methylation status in the DMRs of imprinted genes, MEG3, MAGEL2, SNRPN, NDN, GRB10, and UBE3A, using bisulfite sequencing. The parthenogenetic methylation status of SNRPN, NDN, and GRB10 was maintained after reprogramming and neural differentiation (Fig. 2). SNRPN and NDN are located in the Prader-Willi syndrome (PWS) locus (15q11–q13), and their hyper-methylation status on the maternal allele has been previously reported. SNRPN was hyper-methylated on the maternal allele in iPSCs and differentiated neurons generated from a PWS patient who had only maternal alleles at the PWS locus (17). NDN was also hyper-methylated on the maternal allele in the brain of a patient who had a deleted paternal allele in 15q11–q13 (18). The results of bisulfite sequencing of SNRPN and NDN of parthenogenetic cell lines showed that the hyper-methylated pattern on the maternal allele in the PWS-related region was maintained even after terminal differentiation into neurons and astrocytes (Fig. 2).

Fig. 2
DNA methylation patterns of imprinted genes. Methylation of imprinted genes was analyzed by bisulfite sequencing. Paternally imprinted gene, MEG3; Maternally imprinted genes, MAGEL2, SNRPN, NDN and GRB10; the imprinted gene which have unmethylated CpGs, UBE3A. White and black circles indicate unmethylated and methylated CpG islands, respectively.
![]()
DNA methylation was also maintained in GRB10, which is expressed in a tissue-specific and cell type-specific manner (19). GRB10 has an imprinted region in its brain-specific promoter, which is hyper-methylated on maternal alleles (20). Consistent with these data, we observed the hyper-methylated status of the brain-specific promoter, which activates transcription of the un2 exon of GRB10, in the parthenogenetic cell lines, whereas all of the somatic cell lines showed a hemi-methylated status during reprogramming and differentiation (Fig. 2). The hyper-methylation of the maternal allele and the hypo-methylation of the paternal allele were maintained even after terminal differentiation into neurons and astrocytes.
We found alterations of DNA methylation in MEG3 and MAGEL2 during reprogramming and neural differentiation. MEG3 is a large noncoding RNA acting as a tumor suppressor (21). The DNA methylation status of MEG3 in parthenogenetic cell lines changed from hypo-methylation to hyper-methylation after reprogramming; hyper-methylation was maintained after neural differentiation (Fig. 2). Interestingly, this change was also observed during reprogramming of somatic fibroblasts into hSiPSCs (Fig. 2). According to Nishino et al., the same region of MEG3 was aberrantly hyper-methylated after reprogramming into iPSC lines in various cell types (22). Thus, our data can be interpreted as indicating that aberrant methylation after reprogramming occurred on the maternal allele. In addition, we found that the methylation status of pluripotent stem cells altered during reprogramming was maintained even after differentiation into neural cell lineage. In other words, when aberrant methylation occurred after reprogramming, the methylation status was maintained even after differentiation into neural cell lineage. DNA methylation at CpG islands in MAGEL2, which is a paternally expressed gene closely associated with nervous system development during embryogenesis (23), was altered after reprogramming into hPgi-PSCs. Interestingly, while all somatic cell lines showed obvious hemi-methylation patterns in this region of MAGEL2, the hyper-methylated status in human parthenogenetic fibroblasts changed to a hypo-methylated pattern after reprogramming into hPgiPSCs (Fig. 2). This alteration may be caused by gaining pluripotency. In several imprinted genes, the methylation status is altered after reprogramming by global demethylation during embryo development (24). Thus, the alteration of DNA methylation in DMRs of MAGEL2 including analyzed CpGs could be interpreted as one of the changes during reprogramming that occur on the maternal allele. In addition, hyper-methylation of MAGEL2 on the maternal allele may be considered as a necessary condition for normal nervous system development. Analysis of the Magel2 region in whole-brain genomic DNA showed that most of the CpG islands were hyper-methylated on the maternal allele (25). Possibly, the changes in MAGEL2 DNA methylation suggest that the functional role of MAGEL2 in neural development requires hyper-methylation on the maternal allele during neural differentiation.
We confirmed the DNA methylation status of UBE3A, which is related to Angelman syndrome (AS). As expected, UBE3A was hypo-methylated in both somatic and parthenogenetic cell lines (Fig. 2), consistent with a previous report that UBE3A is not imprinted in all cell types (4).
Our analysis of DNA methylation status in somatic and parthenogenetic cell lines indicated that a methylated imprinting pattern on a maternal allele can be maintained or altered during reprogramming. In addition, we found that the DNA methylation status during neural differentiation from pluripotent stem cells in vitro resembled it in vivo. Therefore, these results support the use of hPgiPSCs as a human in vitro model to study DNA methylation of imprinted genes that is established in a parent-of-origin-dependent manner.
Imprinted gene expression during neural differentiation
Because DNA methylation is one of the mechanisms that control gene expression (26), DMR methylation may be directly related to the expression of some imprinted genes. To assess the potential changes in the expression of imprinted genes during neural differentiation, we performed quantitative real-time PCR in hSiPSCs, hPgiPSCs, and cells differentiated from them.
MEG3 is a maternally expressed imprinted gene on chromosome 14 (21). We observed a higher expression of MEG3 in all parthenogenetic cell lines (hPgiPSCs, hPgi-NSCs, and differentiated neurons and astrocytes) than in hSiPSCs (Fig. 3A). Interestingly, although the CpGs in the analyzed region of MEG3 were hyper-methylated in all parthenogenetic cells (shown in Fig. 2), the expression of MEG3 was detected. Somatic cell lines revealed an increase of the expression of MEG3 after neural differentiation, although an aberrant methylation at hSiPSCs was maintained after neural differentiation. Previously, it was reported that the DMR of MEG3 is aberrantly hyper-methylated and MEG3 is expressed or silenced in several pluripotent stem cell lines (22). Thus, there may be no correlation between MEG3 expression and DNA methylation in analyzed MEG-DMR. We also observed intriguing expression patterns during neural differentiation. While there were no significant differences hSi-NSCs and hPgi-NSCs (t-test, p=0.4105), significant differences were observed between somatic and parthenogenetic iPSCs, neurons, and astrocytes (t-test, p=0.0002; p=0.0009; p= 0.0004, respectively). It suggests that maternal allelic expression is downregulated and paternal allelic MEG3 response for expression in early neurodevelopment. Indeed, maternal uniparental disomy 14, which results in a deletion of paternal alleles including that of MEG3, leads to prenatal growth failure and macrocephaly in humans (27). It means that the paternal allelic expression of MEG3 was essential to normally develop. Collectively, it suggests that the expression of MEG3 might be contrastively occurred on maternal and paternal allele in early neural development.
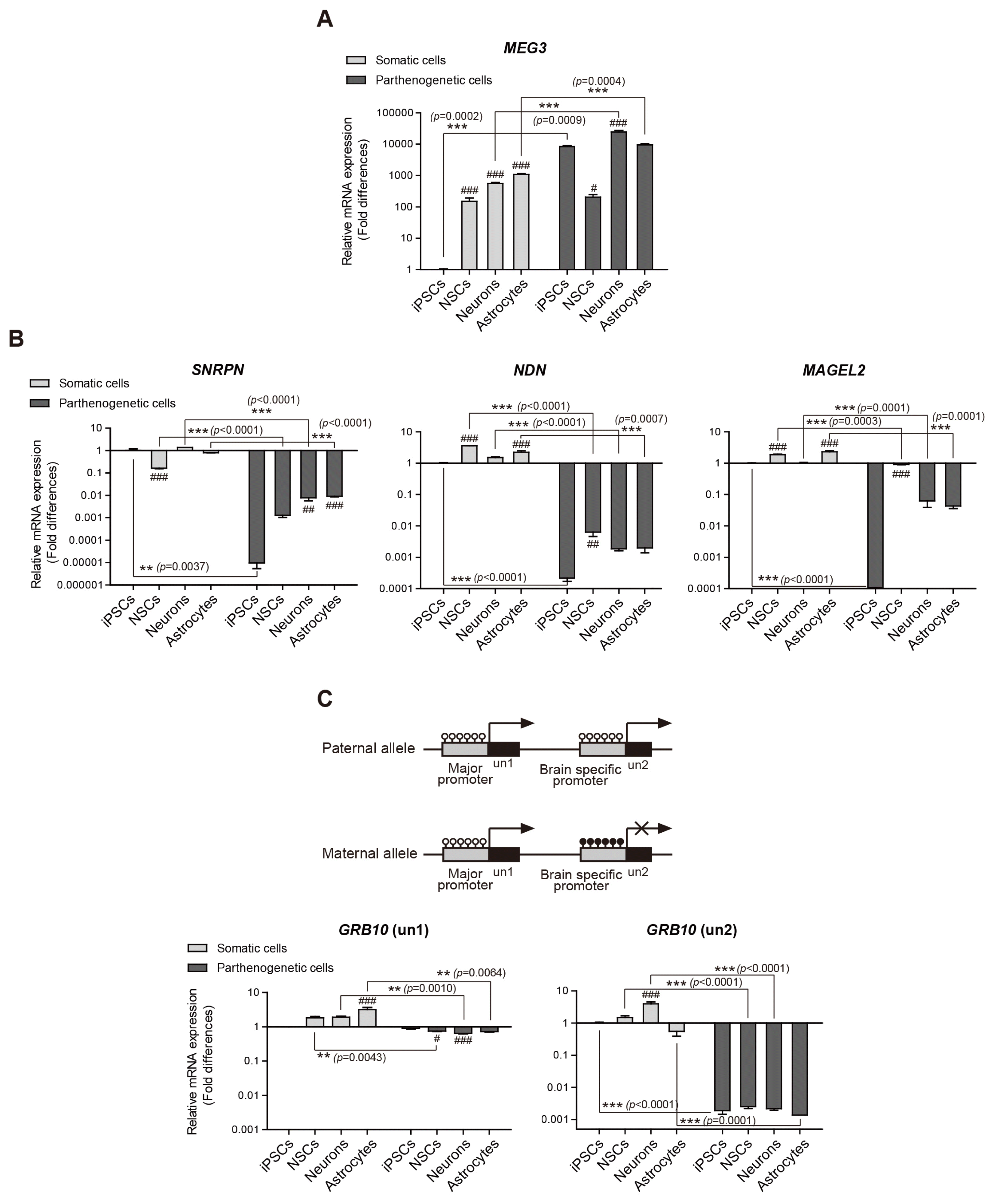
Fig. 3
Expression of MEG3, MAGEL2, SNRPN, NDN, GRB10 (un1), and GRB10 (un2) analyzed using quantitative real-time PCR. The expression levels in hSiPSCs were set to 1. Means±SEM are shown for three independent experiments. Significance of inter-group differences was determined by one-way analysis of variance; #p<0.05, ##p<0.01, ###p<0.001. Significance of the differences between cell types was determined by t-test, and the p-values are indicated each graph with ** and ***.
![]()
We also assessed the expression of SNRPN, NDN, and MAGEL2, which are known as paternally expressed genes. They are located in the PWS region (15q11–q13) and are considered candidate genes for PWS phenotypes (28). As expected, the expression of these genes was not detected in hPgiPSCs (Fig. 3B). However, the expression of SNRPN and MAGEL2 unexpectedly tended to increase as neural differentiation proceeded.
Especially in the case of MAGEL2, the expression in hPgi-NSCs was highly detected unlike in hPgiPSCs and differentiated neurons and astrocytes from hPgi-NSCs. This result can be interpreted as that the maternal allelic expression of MAGEL2 also takes place in early neurodevelopment or neural progenitor cells. In Magel2+m/−p mice, the histological data showed that the maternal allelic expression of Magel2 was specific to the region of the brain where neural progenitor cells were abundant (29). Collectively, this previous study and our data show that genomic imprinting is flexible during development. Whether changes in genomic imprinting are essential for normal development according to the functional role of the imprinted genes should be verified.
We also examined the expression of GRB10, which is expressed in a tissue-specific and cell type-specific manner (19). GRB10 has major promoter and brain-specific promoter that activated differently by methylation in a tissue-specific manner (Fig. 3C) (30). The major promoter, which is unmethylated on both parental alleles, biallelically activates un1 exon expression in most tissues. In contrast, in the brain, the brain-specific promoter only activates paternal allelic expression of the un2 exon, whereas the maternal allele of the promoter is silenced through full methylation. The expression of the un1 exon was detected in all samples, but there was a significant difference in the expression in astrocyte differentiated from hSi-NSCs compared with in hSiPSCs (p<0.001) (Fig. 3C). These in vitro data suggest that un1 expression can be different in a cell type-specific manner, even though it is expressed on both parental alleles in all cell types. As expected, un2 exon expression was detected in all somatic cell lines (although at different levels) but not in any of the parthenogenetic cell lines. In an animal model, the brain-specific promoter is active only in neurons but not in glial cells (20). Our in vitro data that un2 expression was significantly higher in neurons differentiated from hSi-NSCs than in other somatic cell types (p<0.001) are consistent with the in vivo data of Hikichi et al. (2003). In addition, our data showed that there was no direct association between the methylation status in the brain-specific promoter, which all somatic cell lines including neurons have hemi-methylation status in that region (Fig. 2), and the expression of un2 exon.
Overall, we analyzed the expression of imprinted genes in neurodevelopment in vitro through differentiation into neural cell lineage. It was found that the expression of imprinted genes is depending on parental allelic imprinting or altered in a tissue-specific manner during neural differentiation. Our results are consistent with those of previously reported in vivo studies. Thus, this study shows the potential of this model as a human in vitro model useful for understanding the expression of imprinted genes during neurodevelopment.
Molecular signatures of PWS and AS in parthenogenetic cells
Since most imprinted genes are related to development, the deletion or mutation of these genes leads to genetic disorders. PWS and AS, which occur due to a loss of normal expression in the same region of the proximal long arm of chromosome 15q (15q11–q13, the PWS/AS region), are representative neurogenetic disorders. This region contains several candidate genes whose deletion or mutations affect behavioral phenotypes (Fig. 4A) (31). The genes are controlled by an imprinting center (IC) in the PWS/AS region (25). We checked whether the parthenogenetic cells had only maternal imprinting in IC using methylation-specific PCR analysis of the IC region with primers indicated in Fig. 4A. Both the methylated maternal allele (detected as a 174-bp PCR fragment) and the unmethylated paternal allele (100 bp) were observed in all somatic cell lines, whereas only the methylated maternal allele was amplified from each parthenogenetic cell line (Fig. 4B). The monoallelic methylation patterns in the parthenogenetic cell lines obtained from dermoid cysts are in agreement with the genomic imprinting status of maternal alleles in AS patients (32).
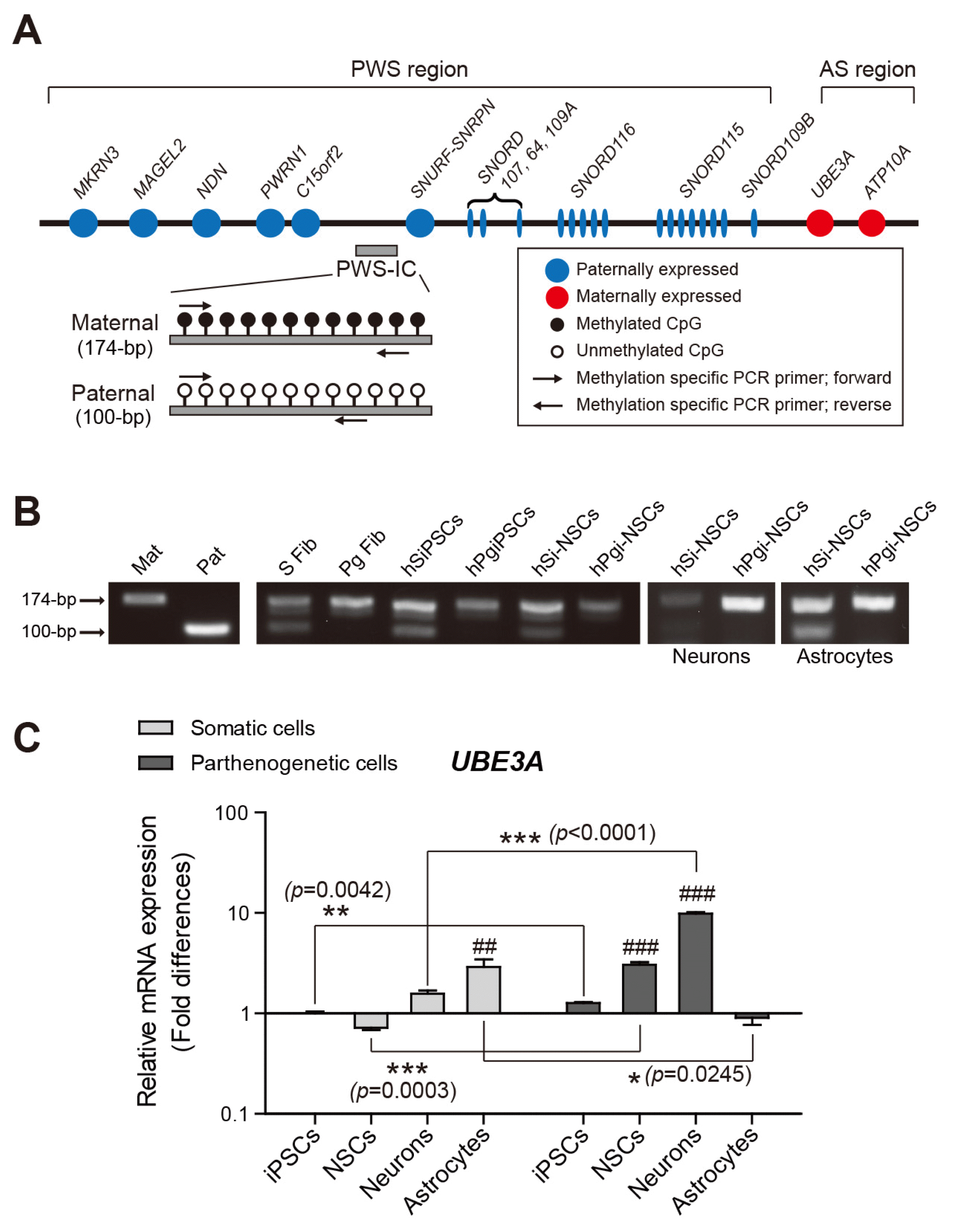
Fig. 4
Imprinting status in the PWS/AS region (Chr. 15q11~13) and UBE3A expression. (A) Schematic representation of the PWS and AS region and primers designed to determine methylation imprints at the IC. PWS: Prader-Willi syndrome, AS: Angelman syndrome, IC: imprinting center. (B) Methylation-specific PCR analysis of the PWS-IC in all somatic and parthenogenetic cells. (C) Quantitative real-time PCR analysis of UBE3A expression in somatic and parthenogenetic cells during neural differentiation. The expression in hSiPSCs was set to 1. Means±SEM are shown for three independent experiments. Significance of inter-group differences was determined by one-way analysis of variance; ##p<0.01, ###p<0.001. Significance of the differences between cell types was determined by t-test, and p-values are indicated with *, **, and ***.
![]()
We also examined whether UBE3A expression is affected by the absence of the paternal allele. UBE3A is located in the AS region of 15q11–q13 and encodes an E3 ubiquitin-protein ligase (33). In neurons, UBE3A is expressed on the maternal allele and is silenced on the paternal allele by activating UBE3A antisense oligonucleotides, whereas the expression in most tissues occurs on both parental alleles (34). UBE3A expression levels significantly changed as neuronal differentiation progressed in hPgi-NSCs, while there were no significant differences during neuronal differentiation of somatic cell lines (Fig. 4C). This result is consistent with the previously reported predominant expression of UBE3A from maternal alleles in neurons differentiated from patient-specific iPSCs (34). Interestingly, however, the expression levels in somatic astrocytes were significantly higher than in parthenogenetic astrocytes (Fig. 4C), although the expression has been reported to be biallelic in astrocytes (35). UBE3A expression in astrocytes has been reported in AS model mice, which had only the paternal allele (36). Despite biallelic UBE3A expression in most cells other than neurons, it can be predicted to occur in astrocytes predominantly on the paternal allele. Our previous study in mice also showed that the expression of Ube3a on the paternal allele was higher in astrocytes differentiated from germline-derived pluripotent stem cell-derived NSCs, which had only the paternal allele, than in astrocytes differentiated from fetal forebrain-derived NSCs (13). Although further studies should be performed, the silenced expression of the gene on the maternal allele in glial cells of AS patients is likely associated with an expression of a protein related to the cellular phenotype of AS.
We established a human in vitro model which could be used to investigate DNA methylation and expression of imprinted genes using uniparental stem cells. Furthermore, we confirmed the maintenance or the alteration of genomic imprinting status of imprinted genes during neural development through in vitro reprogramming and neural lineage differentiation. The genomic imprinting status of the genes analyzed in this study is summarized in Supplementary Table 4. We confirmed that the hPgiPSCs were useful as an alternative source of human uniparental ESCs, which can pose an ethical issue, by examining genomic imprinting status of imprinted genes in various cell types during reprogramming and neural differentiation and confirming the consistence with previous reports. Although there is a limitation for using this model in cell-based therapy because of sex-biased dispersal in all cases of genomic imprinting, it would be suitable for screening new imprinted genes related to neural development. Moreover, the approach reported here can be used for verifying the neuronal cell-specific molecular and cellular phenotypes according to uniparental genotypes.
 XML Download
XML Download