

 PDF
PDF Citation
Citation Print
Print


Introduction
AML is characterized by an abnormal proliferation, without differentiation, of myeloid progenitors, but the biology of the disease is genetically complex and heterogeneous. The clinical course and prognosis in AML vary, depending on the age and category of the disease. AML patients younger than 60 years have complete remission rates of 70–80% (1). However, most of these patients relapse and the 5-year survival rate is less than 45%. Elderly patients have a much worse prognosis with a 5-year survival rate of less than 10% (2). Current chemotherapeutic approaches, which remained unchanged over several decades, have limited effects on the relapsed or refractory AML patients. Thus, novel therapeutics are urgently required to improve AML treatment, and one approach is to find new targets for drug treatment of the disease.
Extracellular signal-regulated kinase 5 (ERK5) is a member of the mitogen-activated protein kinase (MAPK) family, which includes ERK1/2, JNK1/2/3, and p38 isoform (3–5). The ERK5 signaling pathway was the last to be identified and the least studied pathway of the mammalian MAPK cascade. The ERK5 protein has two functional domains that can be dual phosphorylation at TEY motif in N-terminal. It can also be auto-phosphorylated multiple sites in C-terminal, which can function as a transcription factor (6–8). A pharmacological inhibitor, XMD-8-92, is currently available and targets ERK5 pathway activation at different levels. XMD8-92 blocks the C-terminus auto-phosphorylation, resulting in the retention of ERK5 protein in the cytoplasm (9, 10). ERK5 pathway mediates survival, apoptosis, differentiation, and proliferation signaling in embryonic stem cells (11) and several tumor cells including breast cancer, myeloma, lymphoma, leukemia cells (12–16). ERK5 can be phosphorylated as a result of cellular exposure to a broad range of mitogenic stimuli (e.g., growth factors, GPCR agonists, cytokines) and cellular stress (e.g., hypoxia, shear stress) (3–5, 16–20). Although the role of ERK5 has been demonstrated to be required for mitogenic stimuli-induced cell proliferation and cell cycle regulation, its biological significance for the acute myeloid leukemia remains elusive.
In the present study, we provide the evidence that G-CSF (granulocyte colony-stimulating factor) activate ERK5 and that cytokine-induced proliferation is blocked by ERK5 inhibition in AML cells. Furthermore, ERK5 inhibition blocks basal proliferation and has profound anti-leukemic effects leading to cell cycle arrest and apoptosis in AML cells. Subsequent analyses identified c-Myc, hTERT, Cyclin D1, p21, and Bcl-2 as a downstream target affected by ERK5 inhibition. Our study highlights ERK5 as a novel target in AML and supports the use of ERK5 inhibitor as a therapeutic strategy.
Materials and Methods
Cell culture
Human acute myeloid leukemia cell lines, Kasumi-1, HL-60 and MV4-11 cells were purchased from American Type Culture Collection (ATCC). All cell lines were cultured in RPMI1640 supplemented with 10% fetal bovine serum (FBS, Gibco BRL), penicillin (100 units/ml), streptomycin (100 μg/mL) and were maintained at 37°C in a humidified atmosphere of 5% CO2. AML cell lines treated with XMD8-92 (1 and 5 μM) or PD184352 (0.1 and 0.5 μM) were harvested and washed with 1X PBS twice.
Cell proliferation assay
Cells were seeded at a density of 1×105 to 2×105 cells per ml and incubated for 5 hours. Cells were then treated with G-CSF and/or XMD8-92 and PD184352 for the indicated days. The number of viable cells was assessed using the Cell counting kit-8 (CCK-8; Dojindo Laboratoies, Kumamoto, Japan) according to the manufacturer’s instructions.
Cell cycle analysis
Cells were washed twice in PBS and resuspended in 70% ethanol at 4°C overnight. Cells were collected by centrifugation, washed twice in PBS. DNA of cells was stained with 50 μg/ml Propidium iodide (PI) containing 20 units/ml RNase A at 4°C for 30 min. Cell cycle analysis was performed on a FACScalibur flow cytometer (BD Bioscience) equipped with CellQuest Pro software (BD system, San Jose, CA).
Apoptosis analysis
Cells were seeded at a density of 2×105 cells per ml and rested for 4 h. After treatment with G-CSF and/or XMD8-92 or PD184352 for 48 h. The apoptosis analysis was performed using Annexin V-FITC Apoptosis Detection Kit (Abcam, Cambridge, UK), according to the manufacturer’s instructions. Flow cytometry was performed on a FACScalibur equipped with CellQuest Pro software (BD systems). FlowJo software (Version7.6.1, Treestar, Ashland, OR, USA) was used for subsequent analysis.
Western blot analysis
Blots were developed by enhanced chemiluminescence (ECL) according to manufacturer’s instructions (Cell Signaling Tech) and imaged with a LAS-4000 imaging system (Fugifilm Life Science). The following antibodies were used: ERK5 antibody (Abcam), phospho-ERK1/2 antibody (Cell Signaling Technology), ERK1/2 antibody (Cell Signaling Technology), c-Myc antibody (Abcam). β -tubulin antibody (Abcam) was used to normalize the amount of analyzed samples. Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Cell Signaling Technology.
RNA extraction and quantitative Real-time PCR (qRT-PCR)
Total RNA was extracted from cells with the Trizol reagent (Invitrogen), and cDNA was synthesized using High Capacity RNA-to-cDNA kits (Applied Biosystems, Foster City, CA). Real-time PCR was performed in duplicate using SYBR green reagents (Roche, Indianapolis, IN). After pre-amplification (95°C for 2 min), the PCRs amplified for 45 cycles (95°C for 10 sec, 60°C for 10 sec, and 72°C for 10 sec) on LightCycler 96 System (Roche, Indianapolis, IN). Primers used were for c-Myc: F:5′-AATGCAACC TCACAACCTTGGCT-3′ and R:5′-GCCCAAAGTCCAAT TTGAGGCAGT-3′, hTERT: F:5′-GAAAGCCAAGAACGC AGG. GATG-3′ and R:5′-GTCGAGTCAGCTTGAGCAGGAA-3′, Cyclin D1: F:5′-CGCCCTCGGTGTCCTACTTC-3′ and R:5′-CCTCCTCGCACTTCTGTTCCTC-3′, p21: F:5′-GAT GAGTTGGGAGGAGGCAGG-3′ and R:5′-CGAGGCACA AGGGTACAAGACA, Bcl-2: F:5′-CAGGATAACGGAGG CTGGGATG-3′ and R:5′-ACCAGGGCCAAACTGAGCAGAG-3′, and GAPDH: F:5′-CATCAAGAAGGTGGTGAAGC AGG-3′ and R:5′-CGTCAAAGGTGGAGGAGTGGG-3′. The target mRNA expression was quantified using the ΔΔCT method and normalized to GAPDH expression.
Results
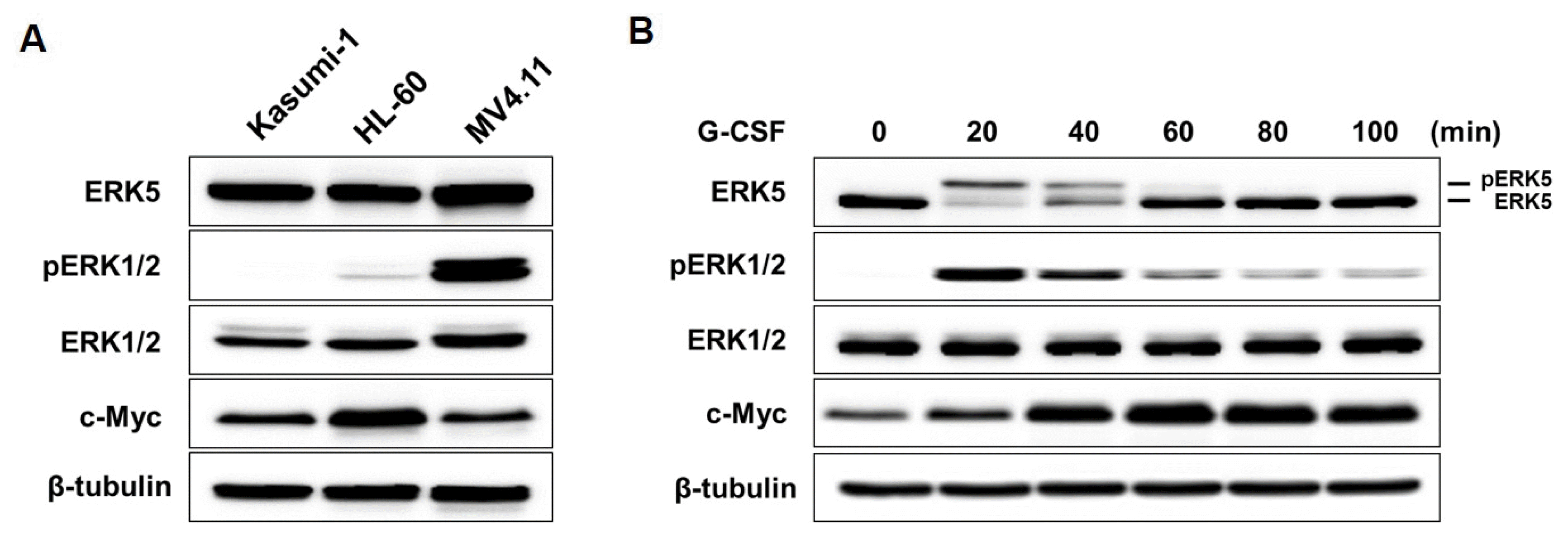
G-CSF induces the activation of ERK5 in AML cells G-CSF, which is the main growth factor for the regulation of myeloid cells proliferation and differentiation, is a common agent used for mobilizing neutrophils, immature myeloid cells and hematopoietic stem cells from the BM (bone marrow) into the peripheral blood (21, 22). Therefore, to consider whether G-CSF treatment enhances ERK5 activity, the activation of ERK5 after treating of Kasumi-1 cells with 10 ng/ml G-CSF was determined. Kasumi-1, HL-60, and MV4;11 cells were very weakly activated ERK5 without G-CSF (Fig. 1A). Kasumi-1 and HL-60 cells were very lowly activated ERK1/2, whereas it was activated in MV4.11 cells. However, ERK5 was activated in Kasumi-1 cells by treatments of G-CSF and induced activation of ERK1/2 (Fig. 1B).
Inhibition of ERK5 by XMD8-92 suppresses proliferation in AML cells
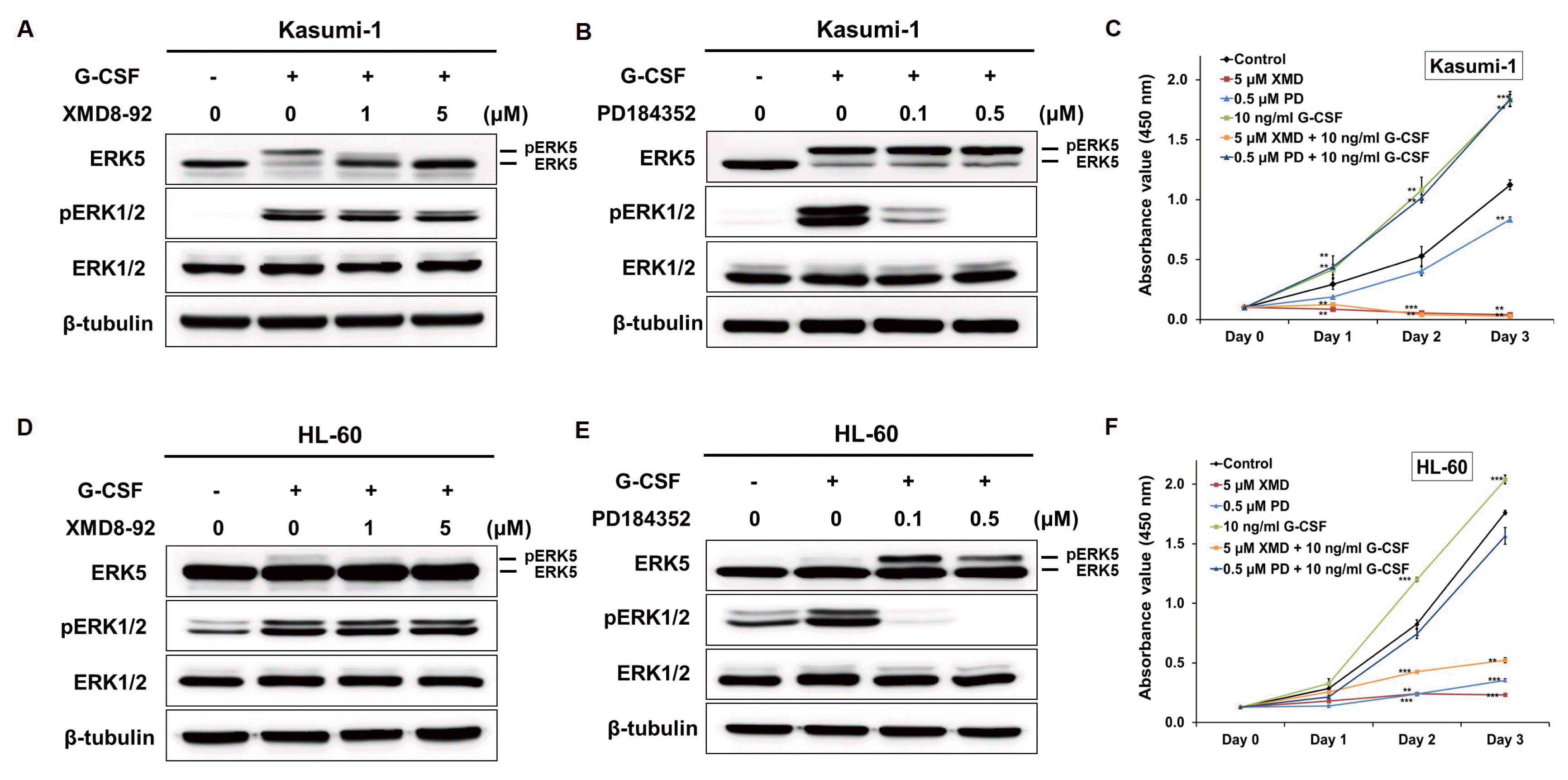
A number of synthetic inhibitors has been used in studies of MAPK functions, and some ERK1/2 inhibitors have been and currently are in clinical trials for various solid tumor and myeloma therapy (23). XMD8-92, a novel ERK5 inhibitor, has been identified for cancer therapy due to its potent anti-cancer activity (24). We tested the effect of XMD8-92 on the cellular activity of ERK5 in Kasumi-1 and HL-60 cells. G-CSF-induced activation of ERK5 and ERK1/2 was effectively inhibited ERK5 by XMD8-92, whereas it did not block ERK1/2 (Fig. 2A and 2D). PD184352, a known ERK1/2 inhibitor, blocked ERK1/2 but did not inhibit ERK5 activation by G-CSF (Fig. 2B and 2E).
Since either the ERK5 and the ERK1/2 pathway was activated by G-CSF treatment, we sought to find which was involved in the proliferation of AML cells. Following the treatment with G-CSF and/or XMD8-92, it was observed that the proliferation of Kasumi-1 and HL-60 cells was increased by G-CSF and suppressed by XMD8-92 (Fig. 2C and 2F). Taken together, these results suggested that ERK5 plays an essential role for the proliferation of Kasumi-1 and HL-60 cells.
Pharmacological inhibition of ERK5 induces cell cycle arrest and apoptosis in AML cells
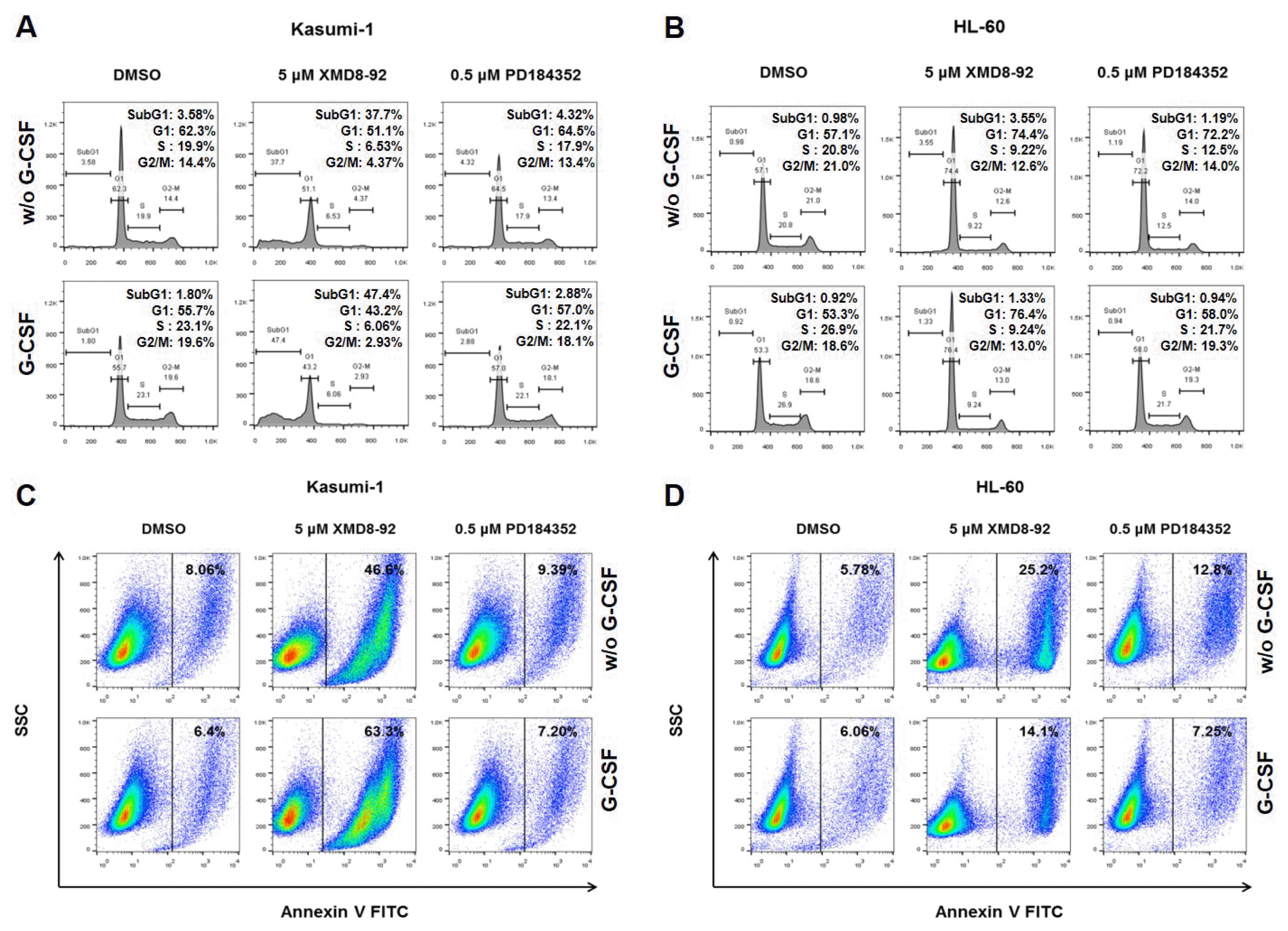
It is known that ERK5 plays a critical role for cell proliferation and apoptosis in several cancer cells (12–16, 25–27). To examine whether the proliferation of ERK5 inhibited cells was due to the disruption of cell cycle phases, flow cytometry analysis was performed on asynchronously proliferating cells with or without G-CSF, XMD8-92, and PD184352. Interestingly, the proportion of cells in sub-G1 phase of XMD8-92 treated Kasumi-1 cells significantly increased 37.7% (without G-CSF) and 47.4% (with G-CSF) to compare with those of untreated (3.58%) and only G-CSF treated (1.80%) Kasumi-1 cells (Fig. 3A). The proportion of cells in the sub-G1 phase of XMD8-92 and G-CSF treated Kasumi-1 cells was more increased in cells without G-CSF. However, XMD-8-92 treated HL-60 cells (without G-CSF: 57.1% and with G-CSF: 53.3%) arrested at G1 phase (without G-CSF: 74.4% and with G-CSF: 76.4%) (Fig. 3B). No significant change was observed in PD184352 treated Kasumi-1 and HL-60 cells with G-CSF (Fig. 3A and 3B). These results indicated that the inhibition of ERK5 by XMD8-92 with G-CSF affected the proliferation of AML cells through the increase of the entry into sub-G1 and G1 phases.
To confirm whether the inhibition of ERK5 induces apoptosis, we labelled XMD8-92 treated cells with Annexin V and analyzed by flow cytometry. Compare with the control Kasumi-1 cells (with or without G-CSF), apoptosis was considerably increased in XMD8-92 treated cells with or without G-CSF (Fig. 3C). The treatment of XMD8-92 in HL-60 cells also induced apoptosis than the control HL-60 cells (Fig. 3D). PD184352 did not induce apoptosis in AML cells with G-CSF (Fig. 3C and 3D). These results showed that the pharmacological inhibition of ERK5 is capable of inducing apoptosis in AML cells.
ERK5 regulates the expression of cell proliferation and anti-apoptotic genes
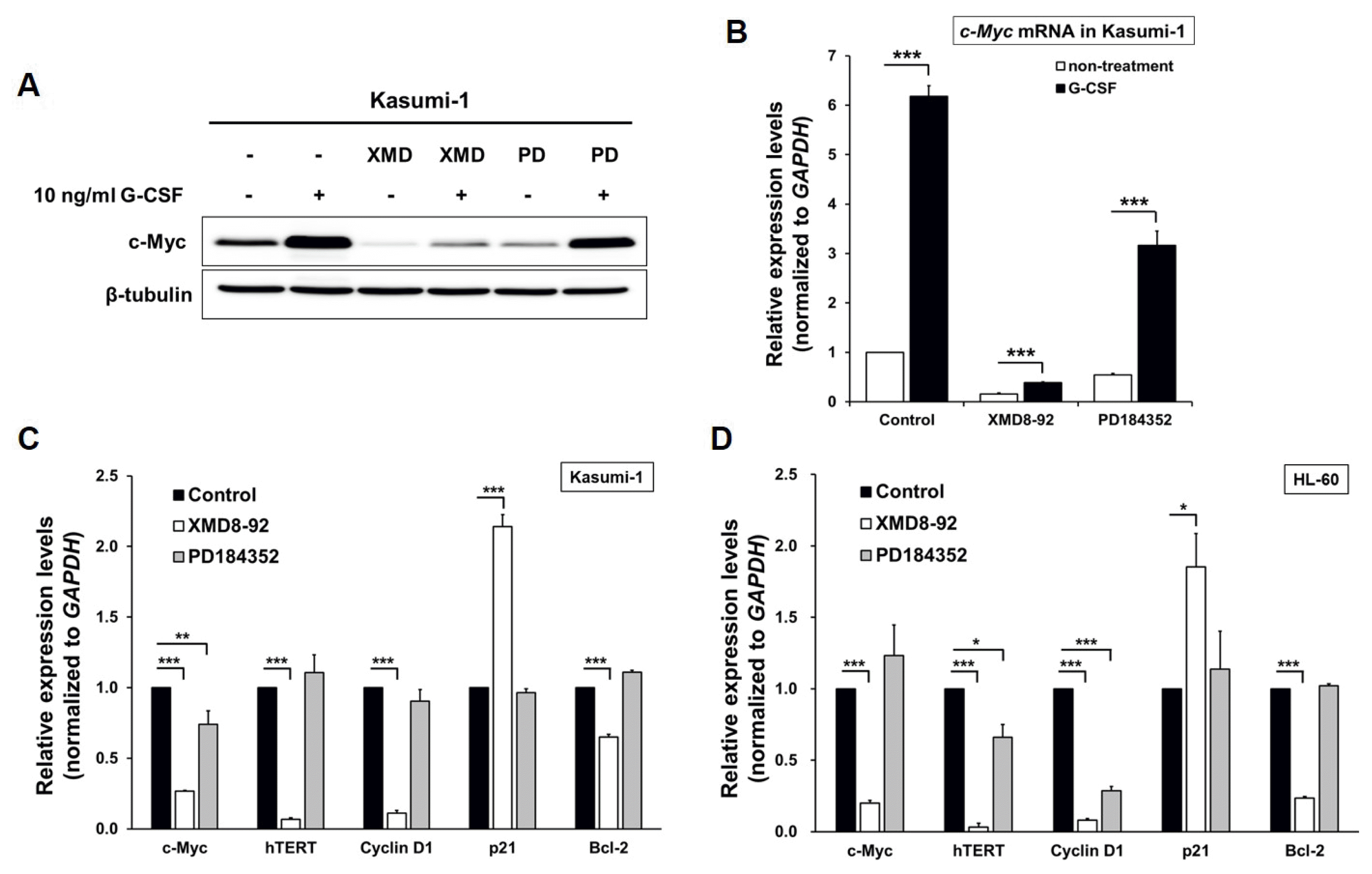
It is reported that the ERK5 pathway play a pivotal role as the mediator of cell proliferation through modulation of cell cycle regulators including c-Myc, c-Fos, cyclin D1, SGK, RSK2 and NF-κB (28–34). To investigate the association between ERK5 and c-Myc, we examined the expression level of c-Myc in the AML cells treated with XMD8-92 by western blot and qRT-PCR. c-Myc expression at both levels was increased by G-CSF and decreased in Kasumi-1 cells treated with XMD8-92 (Fig. 4A and 4B). In addition, we found that the transcripts of hTERT, Cyclin D1, and Bcl-2, as an antiapoptotic gene, was decreased in Kasumi-1 and HL-60 cells through the pharmacological inhibition of ERK5 with XMD8-92 (Fig. 4C and 4D). p21, a regulator of the G1 to S phase transition in the cell cycle, increased in the AML cells treated with XMD8-92 to inhibit ERK5 activity (Fig. 4C and 4D). Together, these results suggest that the activation of ERK5 by G-CSF supports AML cell proliferation through prevention of apoptosis and mediation of cell cycle progress.
Discussion
ERK5 is the largest MAPK expressed ubiquitously in mammalian tissues. The role of ERK5 has been demonstrated to implicate in cell survival, anti-apoptosis, angiogenesis, motility, proliferation, and differentiation in various normal and cancer cells (12–16, 25–27). In multiple myeloma, the inhibition of ERK5 by dexamethasone blocks cell proliferation and induces apoptosis (13). Hodgkin lymphoma and chronic myeloid leukemia show a constitutive activation of the ERK5 (14). In this study, we report that the extracellular signal-regulated kinase 5 (ERK5) is activated in AML cells treated with G-GSF and suppressed by XMD8-92, a pharmacological inhibitor of ERK5. We found that XMD8-92 induced apoptosis and arrests cell cycle at sub G1 and G1 phase in AML cells. Furthermore, we observed that the expression levels of c-Myc, hTERT, Cyclin D1, p21, and Bcl-2 were regulated by the suppression of ERK5 in AML cells treated with XMD8-92.
ERK5 is activated by the upstream kinase MEK5 in response to several growth factors such as granulocyte colony-stimulating factor (G-CSF), nerve growth factor (NGF), fibroblast growth factor (FGF), and platelet derived growth factor (PDGF) as well as shear stress stimulation (19, 20, 35, 36). In present study, we observed the activation of G-CSF-mediated ERK5 in Kasumi-1 and HL-60 cells (Fig. 1B, 2A and 2D), whereas the ERK5 in MV4.11 cells was activated by GM-CSF instead of G-CSF (data not shown). The higher proliferation of AML cells treated with G-CSF may be related with the activated ERK5 (Fig. 2C and 2F). Furthermore, following the inhibition of ERK5 by XMD8-92, not ERK1/2, in AML significantly reduced the AML cell proliferation (Fig. 2C and 2F). These results suggest that the activation of G-CSF-mediated ERK5 may play a role in AML cell proliferation.
Since cell proliferation and apoptosis are usually linked with cell cycle regulator and apoptotic stimuli, we performed a flow cytometry analysis to measure the percentage of cells in the cell cycle phases and apoptosis. The inhibition of ERK5 in Kasumi-1 cells treated with XMD8-92 increased the proportion of sub G1 phase (apoptotic cells), whereas PD184352, an inhibitor of ERK1/2, did not affect the cell cycle regulation (Fig. 3A). In HL-60 cells, the inhibition of G-CSF-mediated ERK5 activation by XMD8-92 induced cell cycle arrest at G1 phase but the cell cycle regulation remained unaffected in PD184352 treated cells (Fig. 3B). Notably, the results of Annexin V assay showed that the pharmacological inhibition of ERK5 significantly facilitated the apoptosis in Kasumi-1 cells compare to HL-60 cells (Fig. 3C and D).
However, ERK5 plays a critical role in cell proliferation, cell cycle, and apoptosis through the regulation of the various downstream targets including kinases (serum/glucocorticoid-regulated kinase (SGK) and RSK2) and transcription factors (c-Myc, c-Fos, and Cyclin D1) (28–32). Bcl-2 has been reported to be a prognostic factor dictating therapeutic outcome in AML (37, 38). The use of small molecules to inhibit both Bcl-2 and MEK leads to the synergistic induction of apoptosis in AML models (39). The induction of apoptosis and a higher proportion of G1 phase requires changes in the expression level of apoptosis- and cell cycle-related genes in Kasumi-1 and HL-60 cells. Therefore, qRT-PCR analysis shown that the expression levels of c-Myc, hTERT, Cyclin D1, and Bcl-2 decreased in AML cells treated with XMD8-92. p21, known as cyclin-dependent kinase inhibitor 1, increased by the treatment of XMD8-92 (Fig. 4C and 4D). Taken together, these data suggest that the regulation of ERK5 by XMD8-92 could be a potential therapeutic target for treating AML.
Recently, this hypothesis was supported by the evidence that the role of ERK5 in chemoresistance in breast cancer cells. Transfection of dominant-negative forms of ERK5 and treatment of apoptotic inducing drugs revealed the anti-apoptotic functions of ERK5 (40). The inhibition of ERK5 reduced cell viability as the cells responded to death receptor-induced apoptosis in leukemic T cells (33).
In addition, it was reported that in vitro treatment of colon cancer cells with 5-fluorouracil (5-FU) and XMD8-92 dramatically increased apoptosis through the stimulation of p53-dependent transcriptional activation of p21, Puma and revealed a reduced tumor mass compare to the treatment of each compound. These reports support that the ERK5 plays a role in chemoresistance and is a major mediator of cell survival and apoptosis in several cancer cells. In summary, the findings of this study provided evidence that the regulation of ERK5 by a pharmacological inhibitor underscores a critical role in the cell proliferation, cell cycle, and apoptosis regulation of AML cells. However, further investigation using in vivo mouse models of AML will be required to confirm the use of XMD8-92 as a potential therapeutic target to treat AML patients.
Acknowledgments
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science & ICT (NRF-2015M3A9C7030091) and by a grant (715003-07) from the Research Center for Production Management and Technical Development for High Quality Livestock Products through Agriculture, Food and Rural Affairs Research Center Support Program, Ministry of Agriculture, Food and Rural Affairs.
 XML Download
XML Download