

 PDF
PDF Citation
Citation Print
Print


Introduction
Regenerative and translational medicine is a rapidly expanding area made possible by the availability of an abundant source of adipose derived stem cells (ADSCs) from lipoaspirates, less abundant bone marrow derived stem cells (BMSCs) and induced pluripotent stem cells (iPSCs). The need for regenerative therapies in osteogenesis and chondrogenesis has increased interest in transdifferentiation of these cells for autologous transplants and therapy development (1). Not surprisingly, neuronal regeneration and repair therapies are of great interest because of its potential to reverse injuries that have severe effects on quality of life (2, 3).
The generation of differentiated neuronal cells from progenitor stem cells has been attempted by a number of researchers over the last decade (4–6). Several have reported the successful passage of ADSCs and BMSCs, in vitro and in vivo, in the presence of simple chemicals and/or growth factors, such as beta-mercaptoethanol (BME) (7, 8), retinoic acid (RA) (9), dimethylsulfoxide (DMSO) and Butylated hydroxianisole (BHA), to rapidly differentiate morphologically toward a neuronal linage. The resultant cell populations have been shown to express morphological and protein surface marker identities consistent with that seen in primary derived neuronal tissue and cultured neuronal cell lines.
BME is a reducing agent and has been shown to be toxic to certain cell types when presented in concentrations higher than the micromolar range (10). The potential for BME to be used as an inducing agent for neuronal differentiation has been studied in a limited capacity (4, 5) and shown to rapidly cause differentiation into cells presenting neuronal-like morphologies within 24 hours of induction (4, 5, 7, 8). Consistent with these morphological changes, BME induced MSCs have been noted to express neuronal specific markers such as Neuron specific enolase (NSE), β-Tubulin 3 (βT3), Glial Fibral Acidic Protein (GFAP), S-100 and Neudesin (NENF) (4, 5, 7, 8).
However, determining the functionality of the produced cells has proved to be much more difficult. The function of BME transdifferentiated cells or the ability of the produced cells to conduct an action potential would prove the cells produced are terminally differentiated neurons. Barnabé et al. (7) conducted patch clamping to detect the Na+ and K+ currents to determine electrophysiological potential, revealing that the produced cells did not show evidence of Na+ or K+ currents nor the ability to fire action potentials.
The characterisation of differentiation by determining the presence of a small number of markers using antibodies in Western blot or immunofluorescence can result in a false impression of the extent of differentiation. By examining the proteome profiles, an unbiased comparative and quantitative measurement of the extent of biological change through the differentiation process can be performed. Thus the aim of this study is the examination of these cells at the proteome level to investigate the changes in protein abundance of differentiating ADSCs in the presence of beta-mercaptoethanol.
Experimental Procedures
Cell culture
This research was approved by the Macquarie University human research ethics committee (Ref #: 5201100385). The procedure described below is adapted from Bunnell et al. (11). Adult ADSCs were derived from abdominal lipoaspirates and subsequent steps were conducted under sterile conditions in a class II laminar flow hood (Clyde-Apac BH2000 series). Lipoaspirates were rinsed twice in Dulbecco’s Modified Eagle’s Medium (D-MEM, Gibco), connective tissue digested with collagenase type 1 (Gibco) for 45 minutes at 37°C before centrifugation at 1600×g for 10 minutes at 4°C to separate adipocytes from the stromal vascular fraction (SVF). The pellet was resuspended in 3 ml of D-MEM and layered on top of 3 ml of Ficoll Paque PLUS (Sigma-Aldrich) to remove red blood cells from the SVF. The resulting purified stromal vascular fraction (SVF) was aliquoted into a T25 culture flask (Nunc) in Delbucco’s modified eagle medium (D-MEM) Glutmax/F12 (Gibco) with 10% Fetal bovine serum (FBS) (Invitrogen) and 1% antibiotics/antimycotics (ABAM) (Invitrogen) and incubated at 37°C at 5% CO2 for 48 hours until ADSCs adhered to the culture flask. Non-adherent cells were eliminated by replacing the media. All isolations were confirmed CD45 negative and CD90 positive (data not shown). ADSCs were passaged 3~5 times by detaching cells with TrypLE Express (Gibco) and before being utilised in differentiation experiments.
Chemical induction for differentiation
Subconfluent ADSCs were washed twice in pre-warmed sterile D-MEM/F12 (Invitrogen). The cells were then cultured for a further 24 hours in a serum-free pre-induction medium consisting of D-MEM/F12 (Invitrogen), Antibiotics-Antimycotics (ABAM, Invitrogen) and 1 mM β-mercaptoethanol (Sigma). The media was then replaced after 24 hrs with the neural inducing media consisting of D-MEM/F12 (Invitrogen), ABAM (Invitrogen) and 10 mM β-mercaptoethanol (Sigma) as per Woodbury et al. (4).
Microscopy
Cell counts
In vitro cell counts were carried out utilising a novel procedure to determine the approximate colony forming units per square millimetre of cells adherent to the culture flask which were induced for differentiation and subsequently utilised for proteomics. A grid of squares 2.5 ×2.5 mm was printed on a transparent laminate and cut to fit outer bottom side of a T175 culture flask (BD Falcon). Ten squares were chosen and cells counted at 100× on an Olympus CK40 inverted microscope and the cell counts from the ten squares were averaged for each flask to find a mean total cfu per square. For the flask total cell population, the averaged cell number was multiplied by 28000 (16squares*10*175 cm) to find the total cell population in the T175 culture flask. To find cfu/mm2 the average cell number from the ten squares were divided by 2.5 mm. At the final time point cells were removed from the culture flask and an aliquot was stained with trypan blue to determine live/dead ratio using a Neubauer chamber. The total cell number data was also utilised in the Bioplex analysis (described below) to determine the amount of cytokines secreted per cell. This was calculated by multiplying the concentration by the total volume of the flask and dividing by the total cell number at the respective time point. Stained cells were visualised on an Olympus IX51 inverted microscope and images captured with the attached Olympus DP70 camera.
Protein Extraction
Culture media was decanted and the cells washed 2~3 times with sterile 1× Phosphate buffered saline (PBS). Cells were harvested by treating cells with 3 ml TrypLE Express (12604 Gibco) for 10~15 minutes at 37°C. Detached cells were rinsed and collected in 10 ml of sterile 1× PBS in a 15 ml falcon tube. Cells were centrifuged at 1000 rcf for 10 minutes to pellet. Supernatant was decanted and the cell pellet was resuspended in 100 μl of 1% SDS and transferred to 0.65 ml eppendorf tube and boiled for 10 minutes to lyse cell pellets. Lysates were centrifuged at 16000 rcf for 10 minutes to pellet debris. Supernatant was then buffer exchanged into 0.1% SDS with SCC Micro-Biospin columns (BioRad).
1D Electrophoresis
Samples were diluted 1:1 with SDS loading buffer (Invitrogen), heated at 95°C for 10 minutes then centrifuged. Samples were then loaded into 4~12% Bis-Tris Criterion gel (BioRad) in XT-MES (BioRad) running buffer then electrophoresed according to the standard product protocol of 160 V for 50 minutes (BioRad). Upon completion of electrophoresis, gels were either used in western blots or fixed and stained with Flamingo fluorescent protein stain (Biorad). Gels were imaged using a PharosFX Plus (Biorad) imager and Quantity One software (BioRad). The gel was then was over stained with Coomassie Blue G stain for visual comparison.
Western Blot
The Western blot method was adapted from Jobbins et al. (12). The membrane was then placed in a solution containing the one of the following primary monoclonal antibodies: mouse anti-human NeuN/Fox3 (M377100 Biosensis 1:5000), mouse anti-human NF200 (M988100 Biosensis 1:500), rabbit anti-human βT3 (ab18207 Abcam 1:1000) or rabbit anti-human GFAP (ab7260 Abcam 1:50000) diluted in PBS respectively and incubated overnight at 4°C on a gentle rocker. Subsequently washed 3 times with PBS and probed with a secondary antibody either and anti-mouse IgG (A4416 Sigma) or anti-rabbit IgG (A4312 Sigma) dependent on the primary probe. Secondary antibodies were peroxidase or alkaline phosphatase conjugated for development with 3, 3-Diaminobenzidine (DAB) (Sigma) or 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium chloride (BCIP/NBT) (Sigma) respectively.
Bioplex
ADSCs and differentiation conditioned media in 500 μl aliquots were collected at time 0min as a control and at 30 min, 1 hr, 3 hrs, 5 hrs, 20 hrs and 24 hrs subsequent to adding the differentiation media and then stored at −80 degC till assay. Concentrations of IL-1ra, IL-1b, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (p70), IL-13, IL-15, IL-17, Eotaxin, FGF basic, G-CSF, GM-CSF, IFN-γ, MCP-1, MIP-1a, MIP-1b, PDGF-bb, RANTES, TNF-α and vEGF were simultaneously evaluated using a commercially available multiplex bead-based sandwich immunoassay kits (Bioplex human 27-plex, M50-0KCAF0Y BioRad Laboratories). Assays were performed according to the manufacturer’s instructions.
iTRAQ
After cell lysis and protein extraction the total of 4 samples for iTRAQ labelling (1~ ADSCs, 2~ 12 hr BME Differentiation hADSC, 3~24 hr BME Differentiation hADSC and 4~ Glioblastoma control [GBCs]) were buffer exchanged in 0.1% SDS using a Tris free Micro Bio-Spin Chromatography Columns (BioRad) and made up to a final concentration of 60 μg/100 μl each. iTRAQ labelling was performed as per manufacturer’s instructions. Following labelling, samples were combined and fractionated by strong-cation exchange (SCX) chromatography with Polysulfoethyl A column. The SCX fractions were injected with an Eksigent Ultra nanoLC system (Eksigent) onto a ProteCol C18 column (SGE) and peptides eluted using a linear gradient at 600 nl/min over 100 minutes. The eluent was subject to positive ion nanoelectrospray analysis in an information dependant acquisition mode (IDA) with a TripleTOF 5600 (AB Sciex). In IDA mode, a TOFMS survey scan was acquired (m/z 400~1,500, 0.25 second), with the ten most intense multiply charged ions (counts >150) in the survey scan sequentially subjected to MS/MS analysis. MS/MS spectra were accumulated for 200 milliseconds in the mass range m/z 100~1500 with the total cycle time 2.3 seconds. MS/MS data were submitted to ProteinPilot V4.0 (AB Sciex) for data processing using Homo sapiens species. Bias correction was selected. The detected protein threshold (unused ProtScore) was set as larger than 1.3 (better than 95% confidence). FDR (False discovery rate) Analysis was selected. A minimum of five peptide cut-off stringency was used to identify proteins. Volcano plots, Gene ontology and Bioplex heat maps were generated using DanteR software (13).
Results
Microscopy
Human ADSCs (hADSCs) were cultured producing a morphologically homogenous culture with cells exhibiting the spindle-fibroblastic form consistent with current literature. The cells were maintained at subconfluency prior to addition of differentiation media containing BME as per Woodbury et al. (4). Fig. 1A~D shows the rate of cellular remodelling over a 24 hour period after the addition of the differentiation media. Basal ADSCs (Fig. 1A) generally grow in a flat, large fibroblastic configuration. Within 3 hours (Fig. 1B) of neuronal induction the morphological changes became evident with a number of cells showing signs of cytoplasmic retraction toward the nucleus of the cell. The now elongated membrane produced a firm and contracted bipolar or multi-polar configuration. At the 12 hour time point (Fig. 1C) the cells morphological changes are ubiquitous across the cultured population with a majority of the cells presenting the retracted cytoplasm and multi-polar shape with evidence of extensions and processes reaching between cells. The cell bodies appear condensed and light refractive compared to the basal hADSCs. At the 24 hour time point prior to harvesting, the cells have a unique morphology compared to the basal state hADSCs with the majority of adherent cells producing polar extensions and processes reaching between cells with some evidence of detachment. In summary these cells morphologically appear to resemble neuronal cells.

Fig. 1
Morphological comparisons between non-induced ADSCs and chemically induced ADSCs. (A) Basal human ADSCs cultured as a control prior to differentiation presents standard flat fibroblastic morphology. (B~D) hADSCs post induction 3 hours, 12 hours and 24 hours with neurodifferentiation media with the progressive structural remodelling over the 24 hour time period. 10×Magnification.
![]()
iTRAQ proteome comparisons of hADSCs, 12 hour, 24 hour differentiated and GBCs control
The digested proteins from each cell line were labelled with the iTRAQ isobaric tags as follows: hADSCs, 12 hour differentiated hADSCs, 24 hour differentiated hADSCs and GBCs labelled with 114, 115, 116 and 117 isobaric tags respectively. The protein fold changes between samples were done comparatively and are relative to a base denominator, the basal hADSCs -114, and all comparisons were made relative to this, i.e. 115 vs 114, 116 vs 114 and 117 vs 114. This was done to elucidate the relative protein fold changes across the captured and labelled proteome of the differentiating cells, determining the up or down regulation of protein species over time during differentiation.
Table 1 summarises the iTRAQ results of basal hADSCs, hADSCs differentiated for 12 hours, hADSCs differentiated for 24 hours and a control GBCs cell line. The summary table shows the upper 99%, 95% and 66% cut off for detected proteins. The upper 95% range was chosen for all data analysis and, within that cutoff, a total of 2,568 proteins consisting 38,786 distinct peptides were identified from 171,862 spectra (Table 1). An average of 5.89 peptides was matched per protein with an average of 13.88% sequence coverage from the total cohort of the detected proteins. The total number of proteins identified by a single peptide match was 980 proteins from the 2,568 identified which is approximately 37% of identified proteins. The analysis cut off removed proteins with below the average peptides matched (i.e. 5 peptides/protein) to increase the robustness of the dataset and the conclusions drawn. A table of all of the above proteins is available in supplementary material 1A. The subsequent cut offs utilised were based on p-value (<0.05) and fold change (log2< −0.2 or 0.2>). These partitions refine the later analyses to statistically significant proteins which have an average of 20 matched peptides per protein. The ProteinPilot group file, the protein summaries and peptide summary (without background corrections) were exported to XML format for further analysis with specified denominators for inter-sample comparisons through the generation of volcano plots and gene ontology graphs.
Table 1
Mass spectrometry iTRAQ protein and peptides counts with relative cutoffs of ADSCs, ADSCs differentiated for 12 hours and 24 hours and glioblastoma cells protein pilot results
![]()
Volcano plots were generated to visualise the up/down regulated proteins, showing p-values versus the log2 protein fold change of each experimental cell line vs. Basal hADSCs of all 2,568 proteins. The quantitation criteria cut off of significant protein fold changes were completed statistically with the students t-test at p-values of <0.05 and log2 fold change cut off of −0.2 or >0.2. This found 2,418 proteins were directly comparable between any two sample types at a time (Fig. 2A~C). The blue nodes outside the horizontal and vertical asymptotes represent the statistically significant changed proteins above >0.2 log fold change up-regulated proteins and the below <0.2 fold change down-regulated proteins. The grey nodes represent the non-significantly changed proteins with a p-value > 0.05 and within the cut off for fold change.
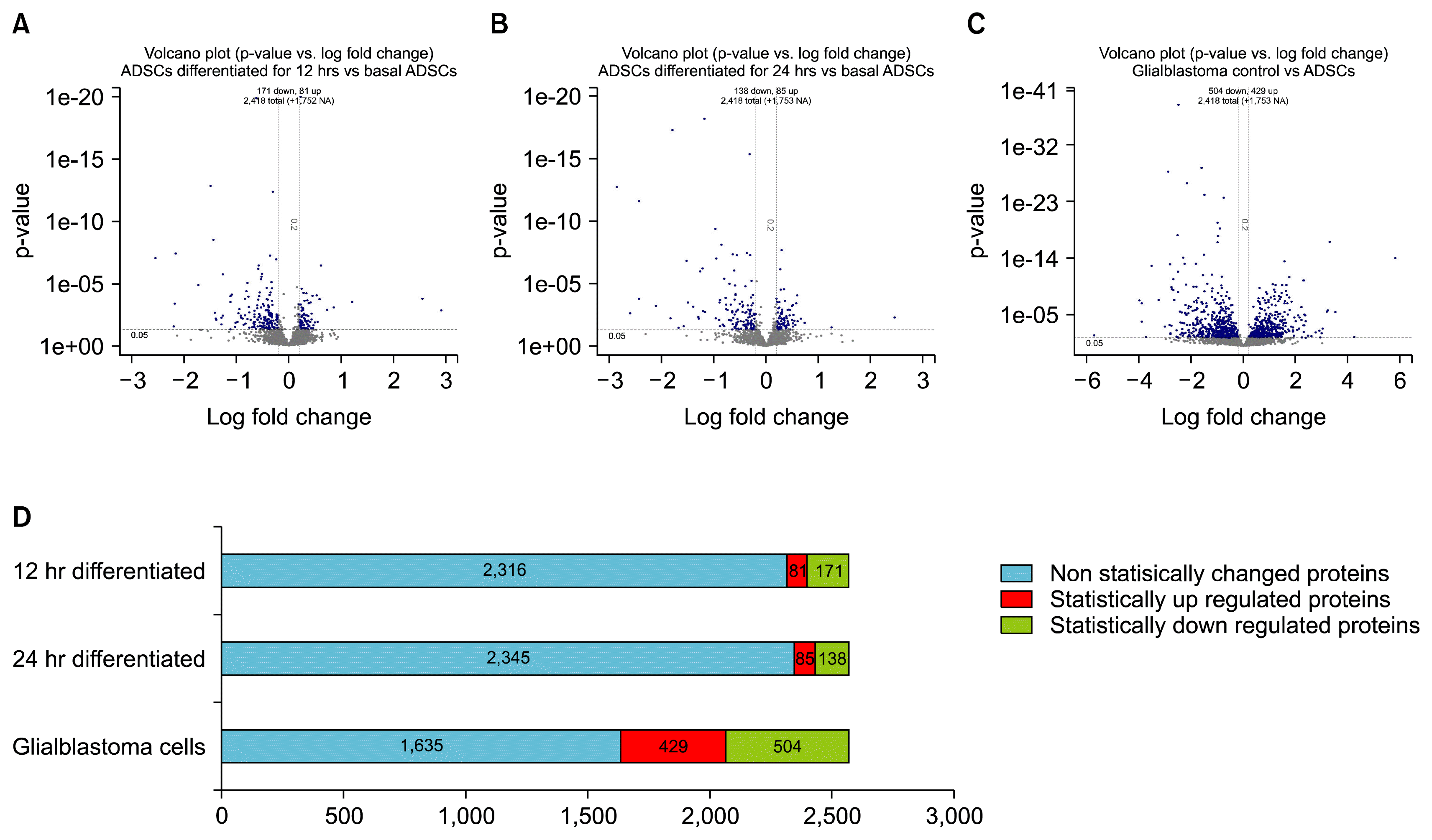
Fig. 2
Volcano plots. (A~C) showing p-values versus protein fold change (log2) of ADSCs and comparisons. Quantitation criteria cutoff of statistically significant p-values <0.05 and log2fold change cutoff of <−0.2 or >0.2. The blue nodes represent the above >0 log fold change i.e. up-regulated proteins and the below <0 fold change down-regulated proteins. The grey nodes represent the not significantly changed proteins with a p-value >0.05 and within the cut off for fold change. D shows the ratio of statistically changed proteins across all samples compared to basal ADSCs. Blue bar presents non-statistically significant changed proteins, red bar is the statistically significant up regulated proteins and green is the statistically significant down regulated proteins.
![]()
The number of statistically significant up and down regulated proteins from each fold comparisons between 12 hr differentiated vs ADSCs (115v114) revealed 81 up regulated and 171 down regulated (Fig. 2A) proteins, comparisons between 24 hr differentiated vs ADSCs (116v114) revealed 85 up regulated and 138 down regulated proteins (Fig. 2B), and comparisons between GBCs vs ADSCs (117v114) revealed 429 up regulated and 504 down regulated proteins (Fig. 2C).
Fig. 2D exhibits the ratio of statistically changed proteins across all samples compared to basal ADSCs in the form of a bar graph. The blue bar presents non-statistically significant changed proteins, red bar is the statistically significant up regulated proteins and green is the statistically significant down regulated proteins.
The Venn diagrams in Fig. 3 presents the statistically relevant up/down regulated proteins that are unique and shared between each time point and the control cell line. Table 2A and 2B presents the up and down regulated proteins respectively. The data listed shows the unique and common proteins to each differentiation time point and their statistical significance as well as fold change relative to the ADSCs. Furthermore the table also shows the relative fold change and p-value of proteins in the other time points to present the extent of change in expression of proteins between the time points. Table 2C presents important proteins related to neurogenesis by cell proliferation, cell differentiation, morphogenesis, cytoskeleton remodelling or response to stress or shock by function according to Gene Ontology biological processes. The mutual expression of neurogenic and stress related proteins indicates the cells are experiencing a directed push toward a phenotype expressing neuronal proteins however the stress proteins indicate that the chemical differentiation is traumatic to the cells and is damaging them throughout the process.
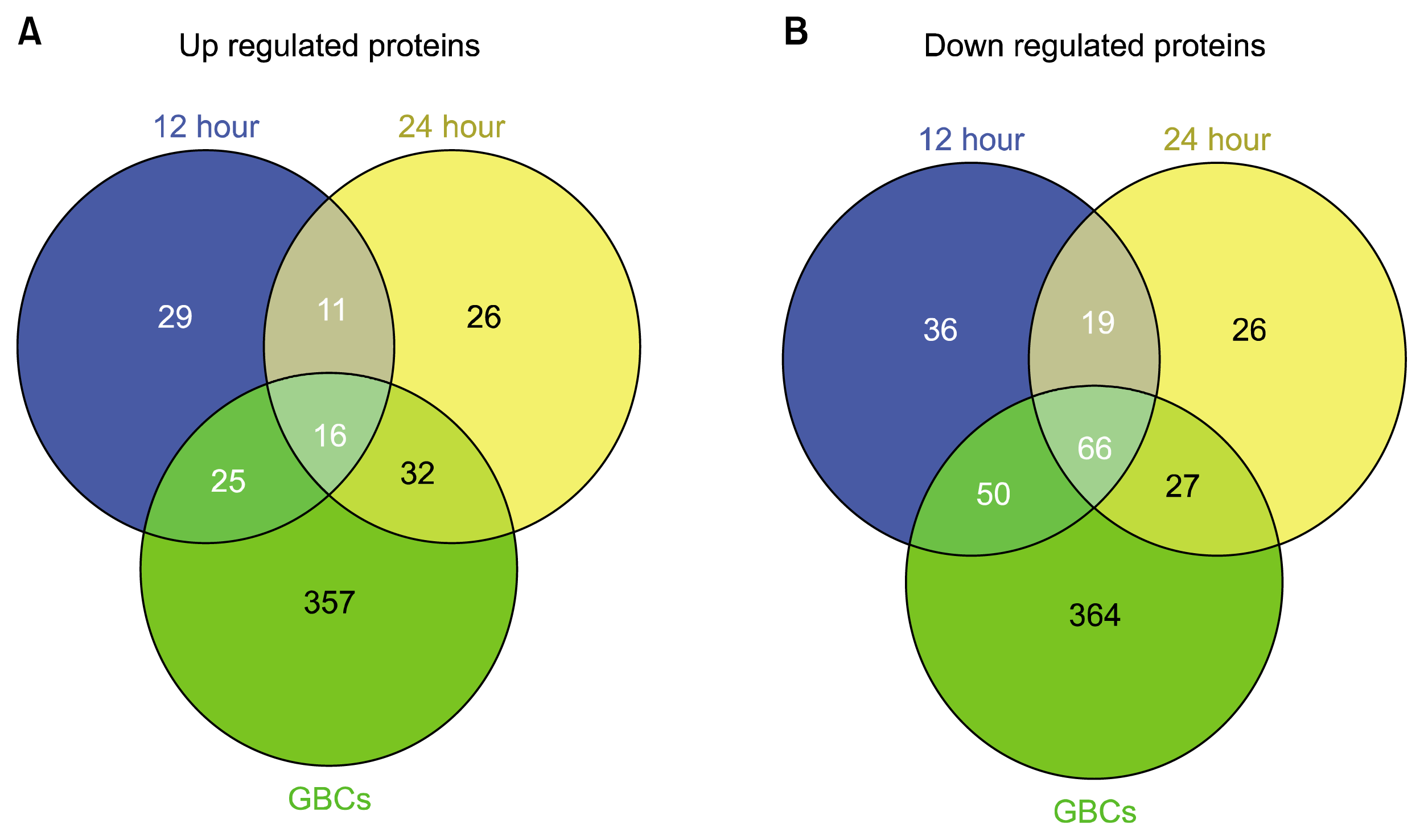
Fig. 3
Three-way Venn diagrams of up and down regulated proteins. Diagrams include the 12 hour differentiated, 24 hour differentiated hADSCs and the GBCs showing unique and shared proteins. (A) shows up regulated proteins revealing there are 29, 26, 357 unique proteins with 11, 32 and 25 shared proteins between each of the corresponding tested cell lines as well as 16 shared proteins between all three relative to basal hADSCs. (B) shows down regulated proteins revealing there are 36, 26, 364 unique proteins with 19, 27 and 50 shared proteins between each of the corresponding tested cell lines as well as 66 shared proteins between all three relative to basal hADSCs.
![]()
Table 2B
Down regulated proteins identified in Volcano plots with fold change and p-values relative to ADSCs
![]()
Table 2C
Statistically significant neuronal and stress proteins expressed by induced stem cells at 12 and 24 hrs
![]()
The down regulation of significant numbers and types of cytoskeletal related proteins, such as actin and tubulin proteins, in the 12 and 24 hr differentiated ADSCs to levels consistent with the GBCs indicates a large morphological restructuring of the cell as identified during microscopy, or damage. The relatively large decrease in myosin related proteins in both differentiation time points indicates that the differentiating cells have shifted away from a mesodermal lineage. This trend is further supported by the mass down regulation of pro-collagen and collagen related structural proteins which, when in high abundance, play a pivotal role in connective tissue, adipose, cartilage and bone formation. Conversely a down regulation of similar structural support proteins is also an indication of cellular damage and stress which has been noted to occur in acute epithelial cell injury (14), and this is covered further in the discussion.
A decrease in the enzyme alpha enolase and the relative increase in gamma enolase/NSE levels are consistent with the development of neuronal tissue seen in rats and humans (15). A switch from alpha enolase, which is mesodermal specific, to gamma enolase which is ectodermal/neuronal specific is often used as an enzymatic biomarker for neuronal development (16). The levels of alpha enolase detected in the differentiation time points are equivalent to the GBC cell line. However, the detected levels of up regulated NSE within the differentiated and GBCs lies in the non-statistically significant identified proteins.
Western Blots
The majority of the aforementioned proteins have more than 5 unique positively matched peptides (Supplementary Table 1). This larger list of neuronal related proteins were deemed to be important and required further investigation as three out of the four most widely used markers for neurogenic differentiation (4–6), Neuron specific enolase (NSE), Neudesin (NENF) and Beta-tubulin III (βT3) fell within this statistically non-significant, up regulated neuronal related protein cut off group. The fulfillment of the 5 peptides or greater cut-off suggests that these molecules are changing but not statistically significantly. Western blots to detect the commonly used neuronal markers βT3, GFAP, NF200 and NeuN were carried out to compare these protein’s expression in the BME treated ADSCs with GBC whole cell lysates in this study and previous literature. βT3 is a 55 kDa protein which is positively detected in the basal ADSCs, the BME differentiated ADSCs and the GBCs (Fig. 4A). The GFAP molecule is only detected at the 48 kDa mark in the GBC lane at a normal exposure (Fig. 4B). By decreasing the contrast by 20%, the GFAP is just detectable in the BME differentiated cells. The NF200 protein is detected in both the ADSC differentiated and with a slightly stronger presence in the GBC at 200 kDa while there is no trace present in the BME lane (Fig. 4C). The NeuN however was not detected in the ADSCs but very faintly in the BME differentiated and GBCs (Fig. 4D). βT3 has been used to characterise primitive neuroepithelium and catalogued as being solely expressed on neuronal cells (17) however βT3 has been found to be expressed in the ADSCs within this study. Similarly the presence of NeuN in the ADSCs confounds the use of this protein as a neuronal specific marker. Thus the identification and quantitatation of the extent of differentiation with relatively few markers is insufficient thus supporting the wider proteomic analysis.
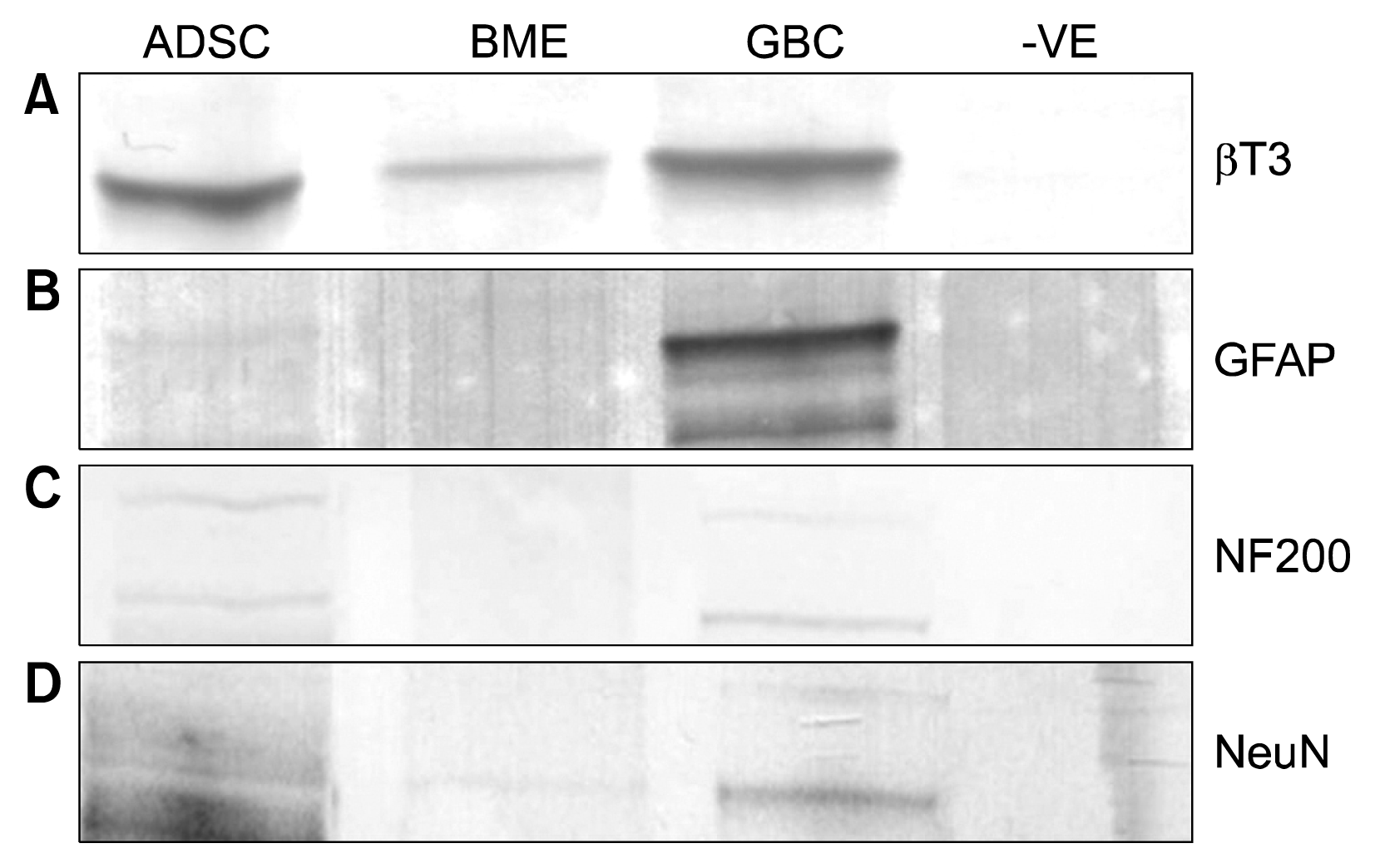
Fig. 4
(A) Western blot of BT3 positive in ADSCs, BME differentiated and GBCs seen at 55 kDa. (B) GFAP positively detected in GBCs at 48 kDa. (C) NF200 positively identified at 200 kDa in GBCs and very weakly in ADSC. (D) NeuN a very low positive in BME differentiated and GBCs. −VE is a negative control of a whole cell lysate of an unrelated cell line.
![]()
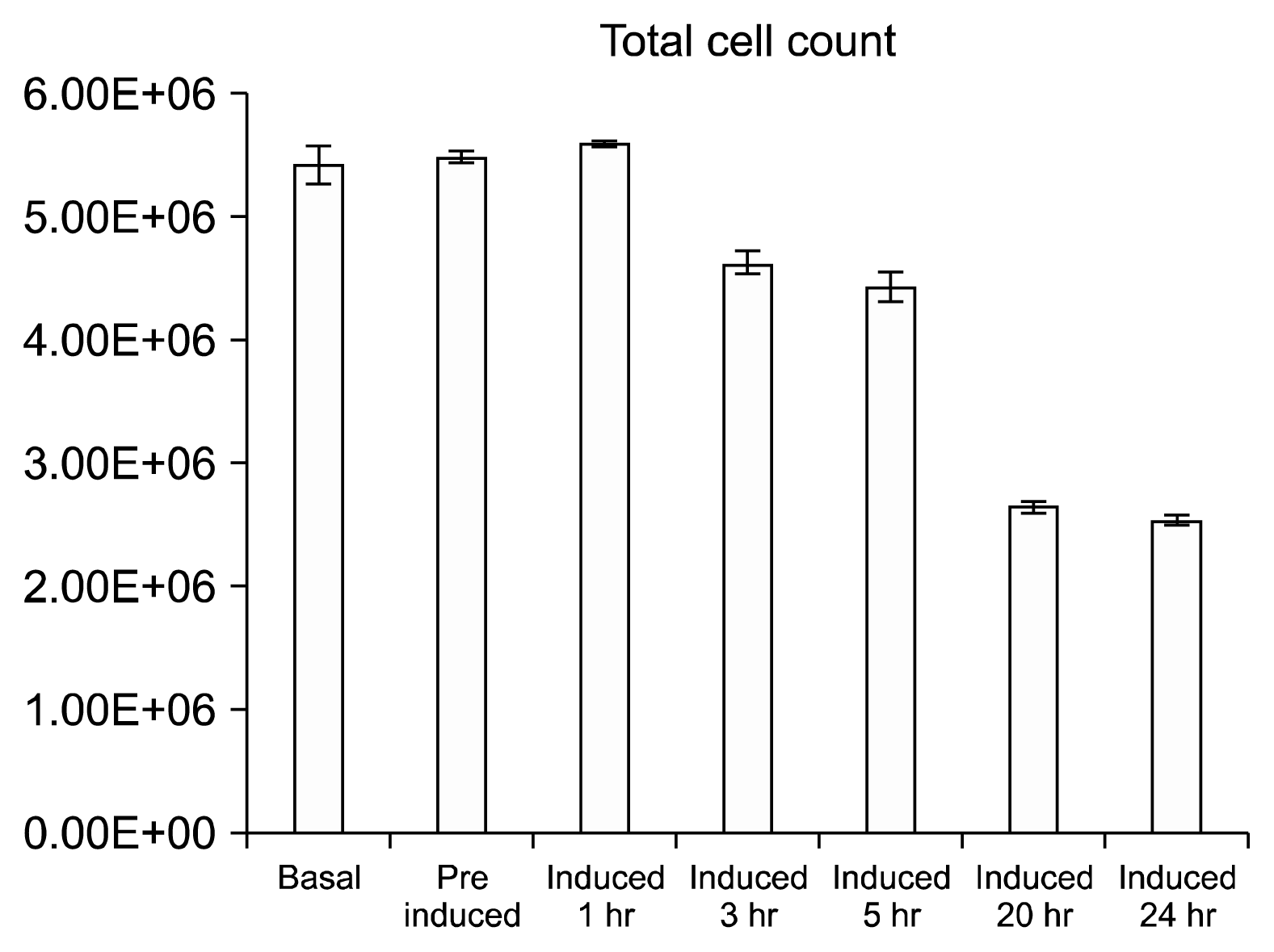
Cell Counts
The total cell count and average cfu/mm2 trends (Fig. 5) are identical presenting no change in cell population in the triplicate flasks from basal cells up to 1 hr post induction. The basal population counts averaged at 77 cfu/mm2 or total population of 5.42E+06 cells. Subsequent to the BME treatment the population decreases by approximately 18% to 63 cfu/mm2 by the 5 hour time point. After 20 hours the cell population decreases by 46%, to 35 cfu/mm2, relative to the basal cells (Fig. 5). Upon harvest of the final time point the total dead/live ratio was 1:9 i.e. an average of 10% of cells were stained blue with trypan.
Bioplex
The Bioplex assay is an efficient system for examining up to 27 cytokines across multiple sample types simultaneously, revealing quantitative changes and relative concentrations of these secreted molecules. Media that the cells were growing in were collected and analysed from the differentiation time points 0, 1, 3, 5, 20 and 24 hours. A hierarchical clustering and Euclidean test in the DanteR software were used to cluster the multiple data points in a heat map configuration where red represents expression above median; green: expression below the median and Black: median expression across all samples (Fig. 6). The hierarchical clustering (Fig. 6) presents the cytokines with similar concentration trends over the differentiation time points.
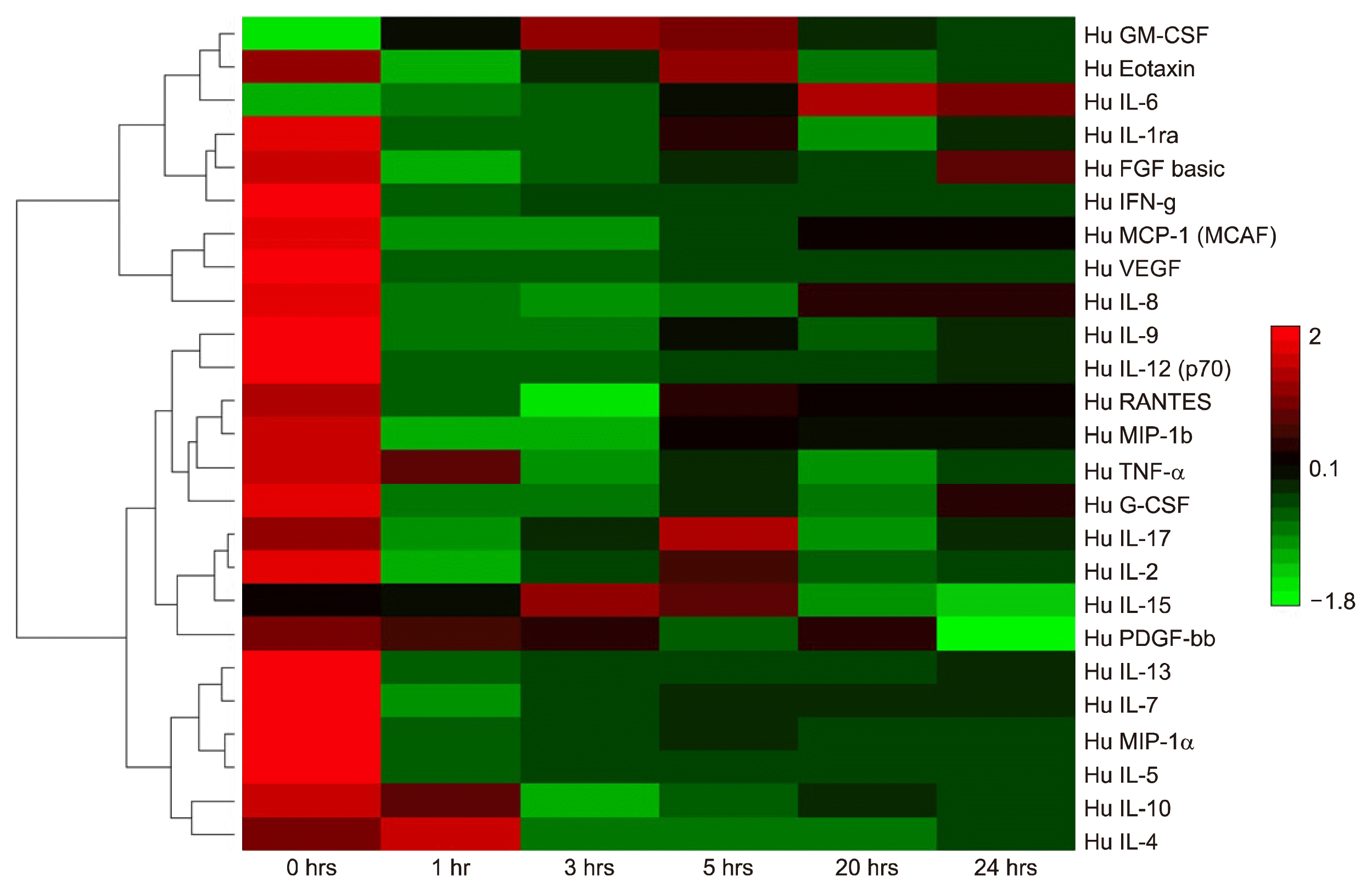
Fig. 6
Bioplex 27-plex cytokine array of ADSCs treated with BME over time. Bioplex comparisons of interleukins and cytokine secretions from basal ADSCs and temporal differentiation with BME neuronal differentiation media. Hierarchical clustering software and Euclidean test Red: expression above median; Green: expression below the median; Black: median expression across sample.
![]()
Cytokines, while having their unique roles in metabolic and cellular processes, can often be regulated in synchrony or regulate the expression of other cytokines in MSCs (18). Individually and collectively their relative concentrations can be related to particular cellular events. As such a number of trends occur within the Bioplex temporal differentiation data set. The molecules IL-1ra, Eotaxin, IL-2 and Rantes share a uniform trend in this dataset with similar concentration fluctuations between IL-1ra and Eotaxin which are comparable to IL-2 and Rantes. The trend reveals the highest concentration of the respective molecules is present at the 0hr time point, with a uniform decrease to the lowest concentrations at the 1 hr time point. This is followed by a slight recovery at the 3rd through the 5th hr. The concentration decreases fractionally again at the 20th hr for IL-1ra and Eotaxin then stabilises at the final time point The next group of trend related cytokines were composed of IL-4, IL5, IL-9, MIP-1a and MIP-1b which followed a fairly simple and distinct trend. The highest concentration occurs at time point 0hrs which then decreases by approximately 75% for all five cytokines thereafter and remains comparatively at the similar concentrations for all time points, with a minor recovery at 3 hrs post induction (Supplementary Table 2). The group consisting of IL-7, IL-13, PDGF-bb, TNF-a, MCP, IFN-g have a somewhat similar trend to the previous group, in that the highest concentrations occur at time point 0hrs, the major defining trend for this series is the significant decrease in concentration to less than 10% in the next time point which is maintained over the course of the differentiation time showing a marginal increase at the 24 hour mark (Supplementary Table 2). The following group: IL-8, IL-10, IL-12, G-CSF and VEG-F also share some common features with the previous two groups in that the highest concentration is observable at time point 0 hrs. The difference is the substantial decrease to near non-detectable concentrations for the entirety of the differentiation (Supplementary Table 2). Conversely the two remaining cytokines IL-6 and FGF were grouped together due to their unique trends that appear to be somewhat related. The main difference between these trends in these two cytokines is that IL-6 appears to have a concentration below the detectable level at time point 0 hrs whereas FGF has approximately 42 pg/ml at the same time point. The trends display a somewhat similar trait after 1 hour with increasing concentrations in both cytokines with the highest levels at the final two time points (Supplementary Table 2).
Discussion
In this study, we investigated hADSCs ability to transdifferentiate toward a neuronal-like lineage within 12 and 24 hours, as well as the changes in the proteome occurring during the differentiation process. It was found that the cells responded to BME differentiation media with similar morphological and marker profiles as previously reported (4, 5, 19) and several groups of proteins involved in neural growth and protection were identified by MS/MS analysis. We also found the acquired soluble proteome of hADSCs differentiated for 12 hours and 24 hours to be noticeably different from basal ADSCs and GBCs, presenting a number of fold changes in proteins related to neuronal differentiation, cytoskeletal remodeling as well as an array of stress response proteins. Furthermore Bioplex cytokine profiles present evidence of a large cellular remodeling shift and stress response activity during the induced differentiation process which is reflected in the proteomic data sets. This study reveals that the BME treatment of ADSCs toward a neurogenic lineage presents a wide array of neuronally related proteins and morphology however the extended exposure of BME to the cells induces a significant stress response.
As demonstrated in this work, the secreted material can be analysed via iTRAQ proteomics or Bioplex cytokine analysis for definitive profiling. Several stress and shock related proteins were identified (Table 2). The proteins Heme oxygenase, Glutamate--cysteine ligase regulatory subunit (GCLM), Glucose-6-phosphate 1-dehydrogenase (G6PD), Prostaglandin G/H synthase 1 (PTGS1), Heat shock protein beta-11 (HSPB11), Stress-70 protein (HSP-70), Keratin, type II cytoskeletal 1, Heat shock protein beta-1 (HSPB1) and 10 kDa heat shock protein (HSPE1), mitochondrial were all investigated to ascertain their role during induction as they were statistically significantly up regulated stress related proteins at the various time points.
The expression of Heme oxygenase (HO) in neurogenic induced ADSCs is an interesting finding since its primary function is the degradation of heme producing biliverdin, iron and carbon monoxide (20). The expression of HO has been found in lung epithelial and liver cell types experiencing oxidative stress (21, 22). Furthermore HO has annotated roles in the response to the action of the oxidative stress linked proinflammitory cytokines IL-1B and TNF-α in astroglial cells (23). It has also been found that HO and the proinflammatory cytokines play a protective role in neuronal cells experiencing oxidative stress as neurons over expressing HO are resistant to oxidative stress mediated cell death (24). The Bioplex results exhibited the expression of IL-1B and TNF-α was far too low for a discernible concentration to be calculated for the former and very low concentrations for the latter to elicit a proinflammatory affect (25). The expression of HO in neurogenic induced ADSCs is undoubtedly due to oxidative stress however the prevention of cell death was not apparent (Fig. 1). Its use as a potential marker indicating cellular distress and protection against oxidative induced death is useful in future studies to indicate stress in chemical inductions (24).
The expression of GCLM has also been linked to a response to oxidative stress; however it has also been detected in high concentrations in muscle cells and lung epithelium undergoing hypoxic or oxidative stress (26). Some studies have linked GCLM to improving the antioxidative defense in astroglial cells by enhancing hydrogen peroxide scavenging ability however this was also in the presence of a thyroid hormone (27), and modulation of the survival of astrocytes and neurons in the presence of reducing agents (28).
Similarly, the high expression of G6PD, like many of the other proteins in this cohort, has been linked to the oxidative stress response by maintaining a redox imbalance-induced apoptosis in a number of cell types (29). Studies in Parkinson’s disease relating oxidative damage to neuronal cells in transgenic mice have shown a moderate increase in G6PD activity and an over expression and neuroprotective activity in aged animals (30).
Equally PTGS1 has been annotated by gene ontology to promote neuronal development and be induced due to oxidative stress. The mechanisms of which PTGS1 is expressed as a neuronal support protein or stress related protein are in response to different signals. The link PTGS1 has to neuronal development is directly associated with the moderate expression of the proinflammatory cytokines IL-1B and TNF-α (31), which in this study has been previously mentioned to have near undetectable concentrations. Studies into their expression during oxidative stress has shown a high expression of PTGS1 from various cell types in the presence of DMSO (32) which incidentally has also been used as an analogous neurogenic induction chemical (3, 4, 7).
Chemically induced oxidative stress to cells has been linked to inhibiting the COX-1 and COX-2 gene which directly affects the proliferation of cells and the high expression of PTGS1 (33). Our findings and the supportive literature indicates that the ADSCs treated with BME neurogenic induction media are responding in a similar manner to other cells types experiencing an overexposure to chemicals which cause an imbalance to the redox state in cells.
Further, to the detected oxidative stress proteins, a variety of additional stress and shock related proteins were noteworthy to this study. HSPB11, which has been linked to an inflammatory response in certain diseases (34), was found to be uniquely and highly expressed in the 24 hour induced ADSCs with none of its ten annotated interacting partners detected in our datasets. Minimal literature is available on the investigation of HSPB11’s role in the stress response. Its presence in the induced ASDCs cannot be accounted for except in response to inflammatory cytokines or due to the oxidative stress experienced by the cells.
HSP-70 is a widely studied heat shock protein and has been found to be ubiquitously expressed in all organisms, linked to the control of cellular proliferation and maintenance during aging (35). One study into homologous stress related proteins in Mytilus edulis have shown a differential in temperature determines the level and sites of expression of stress-70-like proteins in tissue (36). The over expression of HSP-70 has also been linked to the cell’s response to toxic chemicals and the development of cancerous growth (37). The purpose of HSP-70 is postulated to be involved in the protection of cells under thermal or oxidative stress by inhibiting the aggregation of damaged and unfolded proteins (38).
Similarly, HSPB1 and HSPE1 expression is increased when cells are in distress which includes thermal, physical and chemical stressors (Table 2) (39). They are functionally similar to the HSP-70, also annotated in the inhibition of aggregating, damaged or stressed proteins (40). Thus the high expression of heat shock proteins reveals that the ADSCs are being exposed to a prolonged period in traumatic, stress inducing conditions.
Numerous of the above stress related proteins have been linked to the maintenance or protection of neuronal cells experiencing oxidative stress, the remaining shock induced proteins are not related to neuronal cells and are dually expressed due to the extensive stress. While these proteins functions are to preserve the cells in this environment, the loss of approximately 50% of the cell population is alarming and indicates that the surviving cells expressing the detected proteins may in fact be damaged by the induction process.
A shorter treatment may initiate the induction toward a neurogenic differentiation, as an exposure of more than 12 hours to the BME containing media is a stress inducing environment. Our findings essentially indicate that the produced cells may have had the potential for neurogenic differentiation due to the wide variety of neuronal related proteins expressed and detected. However the high abundance of up regulated stress related proteins and high cell death indicates the cells are being damaged. This suggests the culture conditions for inducing ADSCs toward a neurogenic linage with BME is not conducive to producing complete neuronal cells. Nonetheless, the process and mechanisms which drives the cells to differentiate is the most important result acquired as this could be mimicked with much milder and non-toxic chemical cocktails.
Conclusion
The use of chemical inducers to initiate neurogenesis has become well accepted because of its simplicity and due to its relatively rapid outcome of producing morphologically neuronal-like differentiated cells compared to the alternative growth factor induction methods. It is important that the choice of chemical inducer is not toxic to the cells to the extent that major cell death is apparent. While the use of BME produces interesting results in the induction of neurogenesis in ADSCs, it is quite toxic to cells and therefore would not be useful in a clinical setting. This is reflected in decrease in total cell population within the 24 hours of induction. The high death rate in the BME neurogenic induction would not be permissive for in vivo treatments, especially since the extent of damage or stress to the surviving cells has not been well characterised. A catalogue of stress proteins and potential markers can be useful in identifying the biological processes initiated during induced neurogenesis when utilising alternative chemicals to BME.
 XML Download
XML Download