

 PDF
PDF Citation
Citation Print
Print


Introduction
Systemic lupus erythematosus (SLE) is a systemic inflammatory autoimmune disease characterized by a chronic course and variable severity; the management of the disease remains a clinical challenge in patients refractory to conventional treatments (1). In recent decades, cell-based therapies have emerged as a new therapeutic option for refractory and severe SLE in the form of haematopoietic (HSCT) and mesenchymal stromal (MSCs) cells (2–5).
In vitro studies demonstrated imunomodulatory properties of MSCs, thus suggesting their potential use in autoimmune diseases including SLE. The reasons behind the MSC-induced suppression of immunity are complex. MSCs have been found to affect T and B lymphocytes, natural killer and antigen-presenting cells (6).
Moreover, MSCs are an excellent candidate for cell therapy for several reasons: they are easily accessible, they can expand to clinical scales in a relatively short period and, most importantly, allogenic MSCs transplantation does not elicit the host immune-response.
Open-label uncontrolled human trials suggest efficacy of MSCs in treating SLE (3–5); however, although MSCs are considered safe, there are still worries about donor variability, immune-mediated rejection, culture-induced senescence, loss of functional properties, genetic instability or eventual malignant transformation (7–10). Moreover, many issues related the optimal cell source, the most efficient treatment dosage and schedule, the appropriate concomitant treatment, if any remain to be elucidated (11–14). Moreover, in both pre-clinical and clinical studies, a wide range of treatment schedules and administration routes have been tested. In humans, the most adopted MSCs dosage is 1×106 cell/kg of body weight; in animal models doses of 1–2×106 cells have been used, thus a non-comparability of the biological effect between humans and mice could be hypothesized. Moreover, it is well known that high cell doses and higher passage cells can imply deleterious effects on cells survival, engraftment, immunosuppressive functions and lethal thrombotic complications as reported in some animal studies. By using the same MSCs dosage as in humans, we tested the hypothesis that repeated systemic administrations of 1×106 cell/kg of body weight of allogenic MSCs can have a beneficial effect on the course of the disease in a mice model of SLE.
Go to : 
Materials and Methods
All experiments conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and to institutional rules for the care and handling of experimental animals. The protocol was approved by the University of Pisa Ethical Committee.
Animals
The F1 hybrid cross between the New Zealand Bielchowsky Black and New Zealand White mice (NZB/Wf1, subsequently indicated as NZ) (Envigo, Huntingdon, UK) spontaneously develops a disease characterized by immune glomerulonephritis with high levels of antinuclear antibodies and anti-dsDNA antibodies.
Seventy-five 8 weeks old female NZ mice were randomly assigned to receive the following treatments:
Single MSCs infusion at 18 weeks of age (NZs18)
Single MSCs infusion at 22 weeks of age (NZs22)
Multiple monthly MSCs infusions starting at 18 weeks of age (NZM18)
Multiple monthly MSCs infusions starting at 22 weeks of age (NZM22)
Saline infusions (NZc)
In all experimental groups, the treatment (106 MSCs/kg body weight or saline) was provided by caudal vein infusions.
Fifteen 8 weeks old C57BL/6J mice (Envigo, Huntingdon, UK) were used as untreated controls (C).
MSCs isolation and expansion
Bone marrow derived MSCs were obtained from 12-week-old C57BL/6J mice according to a modified version of the Nadri and Soleimani protocol (15, 16). Briefly, the cells suspension recovered from the marrow cavity was gently overlaid on a mouse specific hystopaque (1.083 g/ml, Sigma-Aldrich, St Louis, MO, USA) and centrifuged at 400 g for 30 min, obtaining a visible ring of mononuclear cells, at the interface, that was harvested and washed. The pellet obtained was suspended in a 1 ml volume of DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) for cell count and evaluation of vitality by TrypanBlue (Thermo Fisher Scientific, Waltham, MA, USA) exclusion. The initiating culture cells were seeded at the density of 3×105 cells/cm2. When the cultures reached their confluence, the plates were washed with Phosphate-Buffered Saline without Ca++ and Mg++ (PBS, Thermo Fisher Scientific, Waltham, MA, USA) and incubated for 3 min at 37°C with 1 ml of 0.02% Trypsin/EDTA (Thermo Fisher Scientific, Waltham, MA, USA). Cells were recovered and seeded at a density of 3×103~5×103 cells/cm2 for further expansion. Subsequent passaged cultures were able to reach high level of confluence of monomorphic cells with fibroblastoid spindle-shaped morphology; cells obtained after 7~8 cultural passages were used for the experiments after suspension in PBS.
To confirm the mesenchymal nature of the cells obtained, condrogenic, osteogenic and adipogenic differentiation capability was assessed by adding to the cultures appropriate differentiation media according to the manufacturer’s instructions (StemPro Osteogenesis Differentiation, StemPro Adipogenesis Differentiation and StemPro Chondrogenesis, Thermo Fisher Scientific, Waltham, MA, USA). In addition, surface markers as CD45, CD105, CD106, CD117, CD90 and Sca-1 were evaluated by flow cytometry: cells were washed with PBS, supplemented with 0.1% NaN3 (Sigma-Aldrich, St Louis, MO, USA) and 0.05% bovine serum albumin (BSA, Sigma-Aldrich, St Louis, MO, USA), and incubated with fluorochrome-conjugated monoclonal antibodies as FITC-conjugated anti-mouse CD106, PE-Cy5-conjugated anti-mouse CD117 (1:100, eBioscience, Hatfield UK), PE-conjugated anti-mouse Sca-1, FITC-conjugated anti-mouse CD90.1, PE-conjugated anti-mouse CD105 and PerCP-conjugated anti-mouse CD45, (1:40, Miltenyi Biotec, Bergisch Gladbach, Germany) for 30 min at 4°C, according to the manufacturer’s instructions. After incubation, the cells were washed in PBS/NaN3/BSA and 30,000 events were acquired by MACSQuant cytometer (Miltenyi Biotec, Bergisch Gladbach, Germany) and analyzed by MACS-Quantity software (Miltenyi Biotec, Bergisch Gladbach, Germany). The population of cells with a murine MSC phenotype were Sca-1 positive (>85%) and CD45 negative (<2%) (17).
Assessments of the disease course
Weekly, body weight was recorded and twenty-four hour urines were collected by metabolic cages for each animal; proteinuria was detected by dipstick analysis (Combur Test M; F. Hoffmann-La Roche, Basel, Switzerland).
At sacrifice, peripheral blood samples were collected from mice and anti-dsDNA antibodies were detected by enzyme immunoassorbent assay (ELISA) method using commercial kits (Alpha Diagnostics International, San Antonio, Texas, USA) according to their manufacturer’s instructions. Optical density (OD) at A450 was measured by a microtiter plate reader (Ultrospec2000, Pharmacia Biotech, Piscataway, NJ, USA) and the results reported were expressed as the mean of the optical density readings serum.
Histopathology
Kidneys were excised, fixed overnight in 4% formalin and embedded in paraffin. 1~2 μm sections were cut, dewaxed and stained with hematoxylin and eosin (H&E) (Merck, Kenilworth, NJ, USA) and periodic acid-Schiff (PAS). H&E and PAS stained sections were evaluated for nephritis according to a modified score described by Tao et al. (17).
In detail, glomerulonephritis (proliferative) score was defined as the following: 0=normal glomerulus, 1=focal and mild hypercellularity, 2=multifocal and moderate hypercellularity with capillary dilatation and mild hyalinosis, 3=diffuse hypercellularity (>50% of the tuft) and capillary aneurysm, 4=extensive sclerosis/crescents (>3 cell layer), tuft obliteration, collapse. Interstitial nephritis was defined as: 0=normal tubules and interstitium, 1=small foci of leukocyte infiltration, 2=mild inflammation of individual tubules or isolated atrophied tubules, 3=extensive (>50%) inflammation with large foci of tubular atrophy, 4=nearly all of the interstitium is inflamed+ extensive tubule atrophy or necrosis. Vascular lesions were defined as: 0=no vascular lesions, 1=mild thickening of vessel wall, mild infiltration of leukocytes around vessels, 2=moderate thickening of the vessel wall, inflammation of main arteries, small foci of inflammatory cells around interlobular arteries, 3=severe thickening (onion skin pattern) of vessel walls, moderate leukocyte infiltration of small arterial branches, 4=vasculitis, fibrinoid necrosis
Images were acquired using a AxioImager Z1 microscope (Carl Zeiss MicroImaging, Inc., Jena, Germany). All evaluations were performed in a blinded manner.
Immunohistochemical analysis
Paraffin sections of formalin fixed kidneys were cut (1~2 μm) and incubated with anti-B220 (clone RA3-6B2, eBioscience Hatfield UK) or subjected to a heat-induced epitope retrieval step prior to incubation with primary antibody for CD3 (clone M-20, Santa Cruz Biotechnology, Dallas, Texas, USA), CD4 (clone 4SM95, eBioscience Hatfield UK) or MPO (polyclonal rabbit, Dako Denmark, Glostrup Denmark) followed by incubation with biotinylated rabbit anti-rat, rabbit anti-goat or donkey anti-rabbit (Dianova, Hamburg, Germany). For detection, sections were incubated with alkaline phosphatase (AP) labelled streptavidin (Dako Denmark, Glostrup Denmark). AP was visualized using Fast Red as chromogen (Dako Denmark, Glostrup Denmark). For detection of regulatory T cells, sections stained for CD4 were subjected to protein inactivation step prior to incubation with anti-Foxp3 (clone FJK-16s, eBioscience Hatfield UK). For detection, EnVision+ System- HRP Labelled Polymer Anti-Rabbit (Dako Denmark, Glostrup Denmark) was used. HRP was visualized with diaminobenzidine (Dako Denmark, Glostrup Denmark) as chromogen. Nuclei were counterstained with hematoxylin and slides coverslipped with glycerol gelatine (Merck, Kenilworth, NJ, USA). Negative controls were performed by omitting the primary antibody.
Statistical analysis
Descriptive statistics are displayed as means and standard deviations for continuous variables and proportions for categorical variables. A chi-square test of association was used to compare the distributions of categorical variables between the comparison groups as long as all observed cell counts exceeded 5. Student’s T-test or one way analysis of variance was used to compare continuous characteristics as appropriate followed by Bonferroni post-hoc Test; p-values less than 0.05 were considered statistically significant.
Go to :
Results
Body weight and 24 h proteinuria
NZ mice showed a progressive increase in body weight during the course of the disease with respect to C57BL/6J mice. At all disease stages, there were not weight differences within the NZ subgroups (Table 1).
The curve progress of proteinuria in NZ is detailed in Table 2 and graphically represented in Fig. 1.
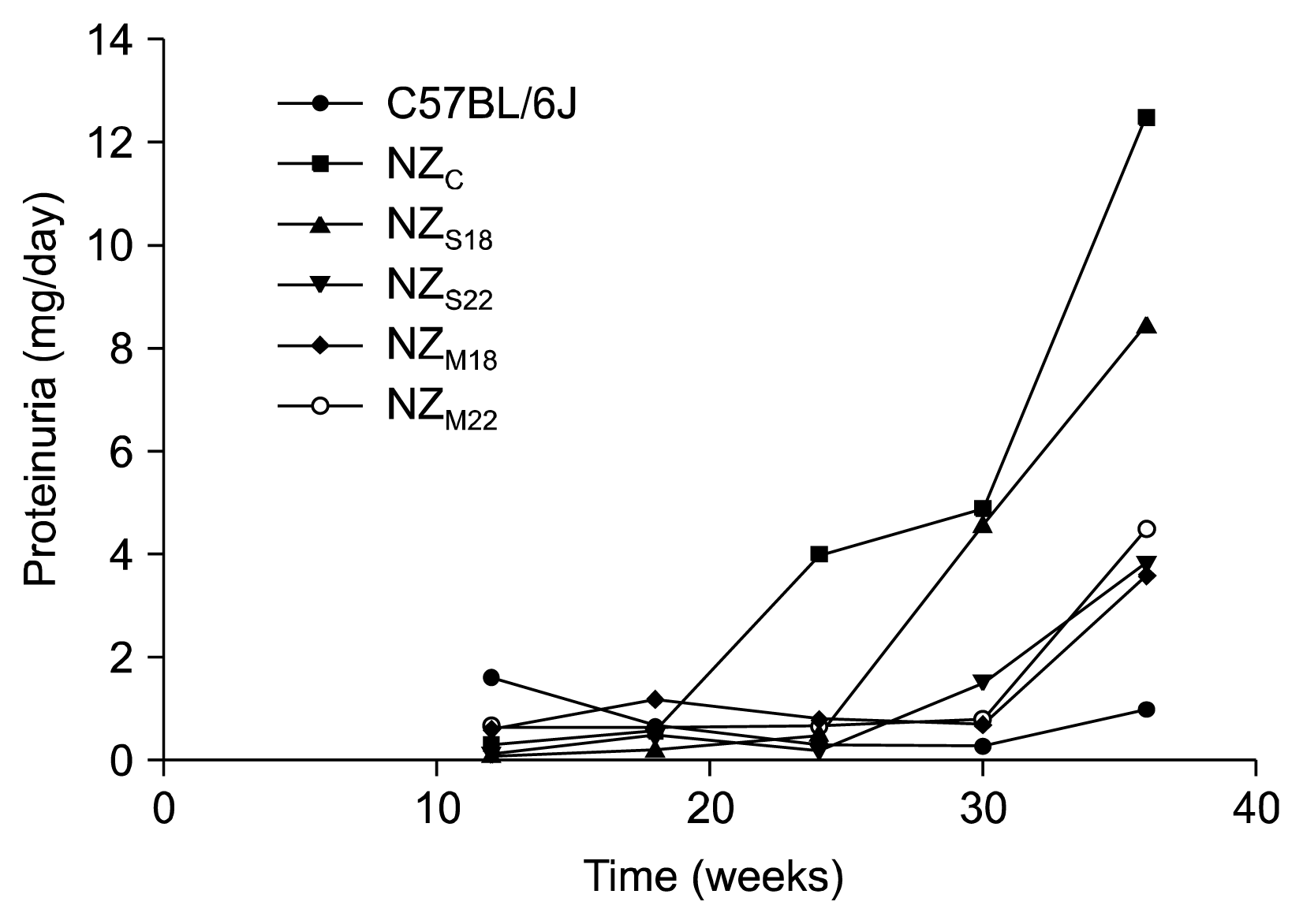
Table 2
Proteinuria (mg/24 hours) in NZ mice subgroups and controls
![]()
In C57BL6/J mice proteinuria was stably below 1 mg/day until 36 weeks of age; in untreated mice (NZc) abnormal proteinuria was firstly detectable at 24 weeks of age (mean 4 mg/day) and further increased until 36 weeks of age (12.5 mg/day).
In mice that received only one MSCs administration (NZs18 and NZs22) proteinuria was first detectable at 30 weeks of age, then the values remained stable until last observation at 36 weeks.
Anti-dsDNA antibody
Anti-dsDNA antibody serum levels for each group over time are showed in Fig. 2.
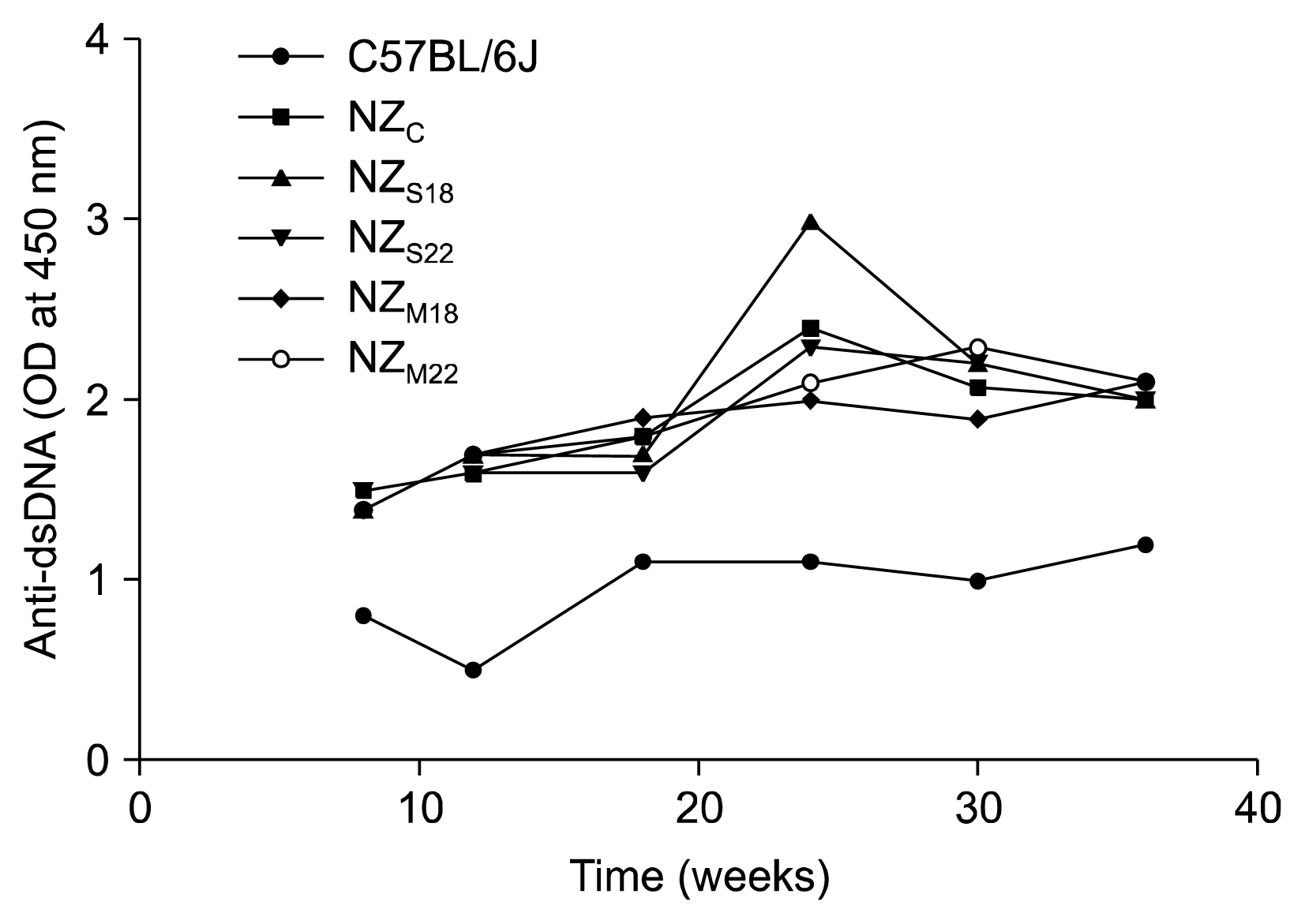
Namely, in NZc, the anti-dsDNA antibodies were detected at 8 weeks (1.5 OD), their mean titer remained stable until week 18 (1.6 at 12 weeks and 1.8 at 18 weeks of age). Thereafter the autoantibodies titer increased and remained stable until final observation (2.4, 2.07 and 2.0 at 24, 30 and 36 weeks of age respectively).
In single and multiple treatment groups, regardless of the time of the starting of treatment (18 weeks or 22 weeks), no differences were observed at the different time points within treatment groups nor with respect to untreated mice.
At the final observation, the mean titer of anti-dsDNA was 2.0 in NZs18, 2.0±0.2 in NZs22 (n.s. versus untreated mice); the mean titer of anti-dsDNA was 2.1 in NZM18, 2.1 in NZM22 group (n.s. versus untreated mice and single treatment mice).
Kidney histology and immunohistochemistry
At the sacrifice in all experimental groups kidneys were examined for histological changes. As expected, no evidence of nephritis was observed in control mice; in NZ, a variable degree of nephritis was observed in all samples; no statistically significant differences were observed between untreated and treated mice (both with single and multiple MSCs infusions).
Results for B220, CD4, CD4Foxp3, F4/80 and MPO are also detailed in Table 3.
Table 3
Kidney histology and immunohistochemistry at 36 weeks of age
![]()
Interestingly, all NZ samples were negative for B220 at 36 weeks, 34% of the NZs samples were positive (the kidneys that displayed B cell infiltration, showed a mild level of infiltration with 82±59 B cells in 10 high power field); on the contrary, all NZm showed a significant B cells infiltration (mean 225 B cells in hpf) (Fig. 3A, Table 3).
 | Fig. 3Kidney Histology and Immunohistochemistry. (A) NZ HE×100 scale bar 100 μm, (B) NZs HE × 100 scale bar 100 μm, (C) NZm HE×100 scale bar 100 μm, (D) NZ CD3-AP B220×100 scale bar 100 μm, (E) NZs CD3-AP B220×100 scale bar 100 μm, (F) NZm CD3-AP B220×100 scale bar 100 μm, (G) NZ CD4-AP Foxp-3×100 scale bar 100 μm, (H) NZs CD4-AP Foxp-3×100 scale bar 100 μm, (I) NZm CD4-AP Foxp-3×100 scale bar 100 μm, (J) NZ MPO-AP×400 scale bar 100 μm, (K) NZs MPO-AP×400 scale bar 100 μm, (L) NZm MPO-AP×400 scale bar 100 μm.
|
No differences were observed in levels of CD4-positive cell infiltration neither between treated and untreated mice, nor within NZs and NZm mice (mean cells/10 hpf in NZ 671.2 vs NZs 389.8 vs NZm 441; n.s.) (Fig. 3B, Table 3). F4/80 is a membrane protein expressed on murine macrophages. No macrophage infiltration was found in kidneys from NZs; while all samples from NZm showed macrophage infiltration with a mean of 206.3 macrophages in 10 hpf; all kidneys from NZ mice showed also infiltration with macrophages though less severe (46 macrophages in 10 hpf) (Fig. 3C, Table 3). Regulatory T cells, as expressed by CD4+Foxp3+ cells, were significantly increased in NZ mice (mean 112.6 cells/hpf) than in NZm (mean 37.2 cells/hpf) while in NZs group no CD4+Foxp3+ cells were found (Fig. 3D, Table 3). MPO staining, as expression of an infiltrate of activated polymorphonuclear neutrophils, was negative in all kidneys from NZ; it was positive in one out of 14 samples in NZs (8%) while the positivity was significantly higher in NZm (85%) with a mean value of 16 (Fig. 3E).
Go to :
Discussion
In this study, we investigated the effect of a systemic treatment with low doses of allogenic MSCs in a spontaneous mouse model of SLE, with special interest on renal findings. The early administration of MSCs (at 18~22 weeks of age), before the development of a full blown clinical disease has been our therapeutic strategy, to evaluate if a timely treatment with MSCs could interfere with the disease development. The effect of single or serial MSCs infusions were also compared.
In fact, we have hypothesized that a treatment with MSCs early after disease onset can modulate the disease process and that low–doses serial administrations can have the advantage of maintaining the beneficial effect over time.
From a clinical point of view, the MSCs administration was associated with a significant delay in proteinuria occurrence, especially in mice that received multiple infusions; however, at our last observation at 36 weeks of age all mice had severe proteinuria with no differences among treated and untreated mice, nor within the treatment subgroups. No effect was observed on autoantibody production by MSCs administration. These data are in line with the histopathological analysis done after sacrifice at 36 weeks of age that did not shown any benefit in term of nephritis scores after treatment with one single or multiple treatments with MSCs. Moreover, an even deleterious effect on inflammatory infiltrate seems to be present in mice that received multiple treatments. Indeed, inflammatory cells deposition (neutrophils, macrophages) in kidneys from mice that received multiple treatments with MSCs is significantly higher than in untreated mice and in mice treated with only one MSCs transplantation. An overexpression of B lymphocytes was also found in mice treated with multiple doses of MSCs while T regulatory cells were reduced in both single and multiple administrations with respect to kidneys from untreated mice. Overall, our study failed to show a positive effect of a treatment with murine MSCs in this model and, for some aspects, even deleterious results seem to be observed.
Previous studies on the therapeutic effect of MSCs in murine lupus models have given encouraging results, although not altogether consistent, mainly for different experimental protocols and the MSCs sources (18–27).
Our study did not fully replicate the positive results recently obtained by Jang et al. that used the same model to evaluate the efficacy of systemic administration of human bone marrow derived MSCs. Indeed, similarly to our results, they showed a delay in proteinuria occurrence in mice treated with MSCs but they also observed a significant delay in autoantibodies production and an improvement in glomerulonephritis. No effect on kidney damage was observed by the same group when MSCs were infused later during the disease, thus suggesting the crucial role of the timing of MSCs administration (22). This aspect was also confirmed by Choi et al who used serial adipose-derived MSCs infusions by comparing the effects in the early versus late disease stage; they found significantly better results when the MSCs administration was initiated before the full-blown disease in term of survival, proteinuria, serological abnormalities as well as in kidney histology (19).
Opposite result was obtained by Youd et al. (25) they used the same NZB/Wf1 model and showed a deleterious effect on lupus disease parameters after intraperitoneal administration of allogenic MSCs at different stages of the disease course. Moreover, anti-dsDNA antibody formation, pathological lesions at kidney histology and immune complex deposition were significantly more pronounced in mice for which MSC treatment was initiated earlier (21 weeks versus 32 weeks of age) and that received the longest treatment course (25). This is in line with our results: even if we cannot demonstrate significant differences in term of clinical and histological parameters in single versus multiple treatments groups, a significant increase on inflammatory infiltrate in kidneys was observed in the latter groups.
Contrasting results were also reported in the experiments by Schena et al. who described an improvement in glomerular proliferation, lymphocytic infiltration and IgG immune complex deposition in kidney from NZB/W mice after serial infusion of allogenic MSCs while no effect on global survival, proteinuria and serum concentrations of anti-dsDNA or ANAs was observed (27).
The discrepancies in the results obtained from different experimental settings highlight the crucial impact that several variables can have on the effectiveness of MSCs as immunosuppressive therapy in these pre-clinical models. The type of SLE model used seems to play a role, since better results have been described when different lupus prone mice were used.
The discrepancy of these pre-clinical data also highlight the importance of a better characterization of the optimal administration strategy to be translated into studies in humans. Moreover, the biological gap between pre-clinical models and human disease seems to be amplified when cell-based therapies are investigated, thus an even more caution when translating data from mice to humans is mandatory.
The therapeutic protocol that we tested in these experiments was built according to some preliminary considerations.
Firstly, our hypothesis was that a MSCs dose in mice similar to what is used so far in the clinical experience could better mimic the MSCs-host interactions; thus, a per-weight adaptation of the human MSCs dosage was used in our mice, thus resulting in a significantly lower dose with respect to previous studies in mice.
Previous studies demonstrated that syngeneic MSCs administration was ineffective in lupus models, probably because of a defective function of MSCs from affected mice (19, 26). Human studies on autologous MSCs transplantation from SLE patients also supported this observation (27–31). Based on these data, we decided to use a different mouse strain as MSCs source. Moreover, even if good results were obtained when mice were infused with human-derived MSCs, we used murine MSCs trying to avoid that inter-order mismatching could affect the results. Bone marrow-derived MSCs are also the better characterized in phenotypes and functions, even if other tissues seem to be promising sources of MSCs for therapeutically use, especially adipose tissue and umbilical cord derived MSCs (5, 32). However, consolidated evidence shows that MSCs from different sources show differences in immunophenotype, proliferation and multilineage differentiation potential (12, 13).
Our study has some limitations; first, the effect of the treatment with MSCs was evaluated by surrogate markers such as proteinuria, anti-dsDNA and kidney histology while no data on mice survival were collected. Moreover, we a priori set up the last observation before sacrifice at 36 weeks of age. Secondly, we use a single model of lupus in our experiments; it could be very interesting to compare the same therapeutic protocols in different models and to evaluate the effect on other disease manifestations.
In conclusion in NZB/Wf1 mice our therapeutic protocol was ineffective in reversing the clinical course of the disease. These data highlight the need of additional investigations to find the optimal therapeutic strategy to be translated in humans. Moreover, because of MSCs multiple origins and heterogeneity, their biology is influenced by a large number of variables most of them unpredictable but potentially affecting their clinical effect (33). Standardization and harmonization of protocols concerning tissue origins, isolating methods, expansion culture, characterization and quality controls are strongly warranted.
Go to :
 XML Download
XML Download