

 PDF
PDF Citation
Citation Print
Print


Introduction
Multipotent stem cells (MSCs) were isolated from adipose tissue for the first time by Zuk and colleagues in 2001 (1). Although MSCs can be found in a variety of tissues fat holds several advantages over others. The frequency of stem cells in fat is 500-fold higher compared to bone marrow (BM), a “golden standard” of adult stem cells (2). Evidence suggests superior proliferative activity of ADSCs over BM stem cells (3). In addition, the procedure of fat collection is relatively easy to perform and less burdensome for donor.
Senescence is metabolically active and stable state of cells both in vitro and in vivo characterized by irreversible cell proliferation arrest and dramatic changes in cell morphology, metabolism, gene expression and secretory phenotype (4).
In 1961, it was discovered that human fibroblasts possessed a limited proliferative capacity in culture, a phenomenon known as replicative senescence (5). DNA of telomeres, terminal structures of chromosomes, shortens during each S phase of cell cycle due to inability of DNA polymerase to complete the replication of lagging DNA strand. Hence, telomere shortening acts as a mitotic clock which determines replicative senescence (6).
Premature senescence, on the other hand, is caused by factors other than critically short telomeres. Among them are the lack of nutrients and cell-to-cell contacts (7), UV radiation (8), reactive oxygen species (9), chemotherapy (10), altered chromatin structure (11), and oncogenes (12).
A variety of biomarkers is studied to characterize MSC senescence. Among them, the most popular ones are associated with morphological and proliferative changes (13), increased expression of senescence-associated β-galactosidase (SA-β-gal) (14), the loss of MSC trilineage differentiation potential (15), cell cycle arrest (16), epigenetic modifications (17), oxidative stress (18), telomere shortening and DNA damage (19), activation of tumor suppressors p53, RB1 and p16INK4 (17, 20, 21). In addition, formation of senescence-associated heterochromatic foci (22) and promyelocytic leukemia protein bodies (23), as well as altered microRNA expression profile (24) have been observed.
A plenitude of clinical data gathered both from animal and human studies suggest broad clinical applicability of MSCs (25, 26). However, a single treatment protocol may require as many as 10~400 million cells to attain clinical significance. Therefore, pre-transplantation cultivation of MSCs is a prerequisite. For this reason, it is essential to study MSC in vitro senescence especially given the lack of standardised MSC expansion protocols among laboratories.
The aim of this work was to analyze in vitro senescence of human culture-expanded ADSCs. Previously frozen and long-term cryopreserved ADSC cultures from 8 donors were cultivated until proliferation arrest was reached. Cell senescence was characterized with respect to cell morphology, proliferative abilities, potential of adipo- and osteogenesis, SA-β-gal expression, metabolic activity, accumulation of intracellular peroxide, transcriptional activity (monitoring of 25 gene expression by reverse transcription polymerase chain reaction (RT-PCR)), as well as relative telomere length.
Materials and Methods
Cell source
ADSC cultures from eight donors were used in the study (Table 1). All cell cultures were retrieved from a cell bank where they were stored at passage 2 (P2) (except for cells from donor CS-5 which were stored at P3). ADSCs were isolated from human subcutaneous adipose tissue in accordance with The Latvian Central Medical Ethics Committee (permit No.12) after informed consent. Primary ADSCs cultures were cultivated in 5% autologous serum in hypoxic conditions (5% O2) and prepared for storage (5×106 cells per ml) as described elsewhere (27).
Table 1
Summary of adipose-derived mesenchymal stem cell cultures used in the study
![]()
Cell culture
After thawing, cells were counted in Bürker chamber and seeded on 75 cm2 tissue culture flasks (~2×105 cells per flask) in DMEM/F12 medium containing 10% fetal bovine serum (FBS), 20 ng/ml basic fibroblast growth factor (bFGF), 2 mM L-glutamine and 100 μ/ml:100 μg/ml penicillin-streptomycin (all reagents were obtained from Life Technologies, Paisley, UK, except for bFGF – BD, Franklin Lakes, New Jersey, USA) and cultured in a humidified atmosphere at +37°C, 5% CO2. Medium was replaced every third day. When ~80~90% confluency was reached cells were detached using cell dissociation reagent (TrypLE Express Enzyme, Life Technologies). A portion of cells were split at a ratio 1:5 for further subculturing. Remaining cells were frozen in DMEM/F12 medium containing 10% DMSO (Gaylord Chemical, Slidell, Louisiana, USA) and 20% FBS and stored in liquid nitrogen for later analysis. Passaging was continued maintaining the 1:5 split ratio until confluency could not be reached within four weeks.
Proliferation kinetics
Cells were counted using Bürker chamber at the end of each passage. Population doubling time (PDT) and the number of population doublings (PD) were calculated according to formulas PDT=ln2*T/ln(NT/N0), and PD=T/PDT, respectively, where T – culture time, NT–cell number at the end of a passage, N0–cell number at the beginning of a passage.
Colony forming unit (CFU) assay
Two replicates of 103 cells were seeded in a six-well culture dish using the same medium as that for cell culture and cultivated for 2 weeks in a humidified atmosphere at +37°C and 5% CO2. Medium was changed every fourth day. Dishes were placed on ice and washed twice with cold phosphate-buffered saline (PBS) (Life Technologies). Cells were fixed with ice-cold methanol for 10 min, removed to room temperature and stained with 0.5% crystal violet dye (Sigma-Aldrich, Steinheim, Germany) solution in methanol. The stain was dissolved with 0.5% sodium dodecyl sulphate and absorbance was measured at 539 nm wave length.
Adipo- and osteogenic differentiation
For adipogenesis, ADSCs were cultivated in 12-well culture dishes until 90% confluent. Regular culture medium was replaced with adipogenesis induction medium containing DMEM with high glucose content (Life Technologies), 10% FBS (Life Technologies), 2 mM L-glutamine (Life Technologies), 10 μg/ml human insulin (Life Technologies), 1 μM dexamethasone (Sigma-Aldrich), 100 μM indomethacin (Sigma-Aldrich), 0.5 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich), 5 μg/ml gentamicin (Life Technologies). Cells in control wells were cultivated in DMEM (high glucose) supplemented with 10% FBS. Cells were cultured in a humidified atmosphere at +37°C and 5% CO2 and medium was replaced every third day. Lipid droplets were detected after 16 days by Oil Red O (Sigma-Aldrich) staining.
For osteogenesis, ADSCs seeded on 12-well plates were allowed to reach 70% confluency. Osteogenic differentiation was induced by adding medium containing DMEM with low glucose content (Life Technologies), 10% FBS (Life Technologies), 2 mM L-glutamine (Life Technologies), 10 mM glycerol-2-phosphate (Sigma-Aldrich), 50 μM L-ascorbic acid (Sigma-Aldrich), 0.1 μM dexamethasone (Sigma-Aldrich), 5 μg/ml gentamicin (Life Technologies). Control cells were cultivated in DMEM (low glucose) with 10% FBS. Cells were cultured in a humidified atmosphere at +37°C and 5% CO2 and medium was replaced every third day. After 30 days, extracellular calcium accumulation was detected by Alizarin Red S (Sigma-Aldrich) staining and alkaline phosphatase activity was determined using BCIP/NBT (Sigma-Aldrich) as a substrate.
SA-β-galactosidase assay
2.5×104 cells were seeded in a 96-well plate in triplicates. After 48 hours, β-gal expression was detected using Senescence Cells Histochemical Staining Kit (Sigma-Aldrich) according to manufacturer’s instructions. To obtain a quantifiable result, images of stained cells were acquired with PowerShot S80 digital camera (Canon, Melville, New York, USA) and evaluated by two independent observers according to two parameters – color intensity and percentage of stained cells per field of view. For color measurement, a 5-point intensity scale was developed where 0 equaled ‘no staining’ and 5 – ‘very dark color’.
MTT assay
2.5×104 cells were seeded in a 96-well plate in triplicates. After 48 hours, MTT assay was performed using Vybrant® MTT Cell Proliferation Assay Kit (Life Technologies) according to manufacturer’s instructions. Absorbance was measured at 539 nm after overnight dissolution of formazan.
Quantitative peroxide assay
100 μl of ADSCs (1×106 cells per ml) were lysed by repeated freezing in liquid nitrogen and thawing at 42°C. Peroxide level in cell lysates was determined using Pierce™ Quantitative Peroxide Assay Kit (Thermo Scientific, Rockford, Illinois, USA) according to manufacturer’s instructions on lipid-compatible procedure. Absorbance was measured at 620 nm.
Reverse transcriptase polymerase chain reaction
RNA was isolated from ADSCs with TRI reagent (Sigma-Aldrich) according to manufacturer’s instructions except that RNA was precipitated in isopropanol overnight at −20°C in the presence of 120 ng of glycogen (Thermo Scientific). RNA quality and quantity was determined by NanoDrop® ND-1000 spectrophotometer (Thermo Scientific). 500 ng of DNase-treated RNA was subjected to complementary DNA synthesis using RevertAid™ First Strand cDNA Synthesis Kit (Thermo Scientific). The reaction was carried out with oligo(dT) primer according to manufacturer’s instructions. 1 μl of cDNA was added to a PCR reaction mixture containing 2X PCR Master Mix (Thermo Scientific) and 0.4 μM primers (Metabion, Planegg, Germany) to amplify a set of genes (Table S1). Reaction conditions were as follows: initial denaturation at 94°C for 3 min followed by 30 cycles of 94°C for 30 s, 60°C for 30 s and 72°C for 45 s, finished with a 5 min extension at 72°C. Amplification products were visualized by ethidium bromide staining in agarose gel electrophoresis. Densitometric analysis of acquired images was carried out using ImageJ software (version 1.48).
Telomere Flow FISH
Relative telomere length of cultured ADSCs was assessed with Telomere PNA Kit/FITC for Flow Cytometry (Dako, Glostrup, Denmark) according to manufacturer’s instructions using BD FACSCanto™ flow cytometer with BD FACSDiva™ Software (version 7.0). Jurkat cell line (ATCC/LGC Standards, Boras, Sweden) served as inner control. Each sample was assayed twice. 20 000 events were counted per measurement. Flow cytometer calibration was performed using BD Cytometer Setup and Tracking Beads. Results were analyzed using Infinicyt™ software (version 1.5.0) (Cytognos, Salamanca, Spain).
DNA index
DNA index of ADSC and Jurkat cells was measured with CycleTEST™ PLUS DNA Reagent Kit (BD) using BD FACSCanto™ flow cytometer and ModFit LT v3.3 software (version 3.3) (BD). The procedure was carried out on freshly thawed cells according to manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed in R software (version 3.0.2). Comparison of two data sets was done by t-test or Wilcoxon’s test based on homogeneity of dispersions which was determined by F-test. Relation between results obtained by two different tests was assessed by correlation or regression analysis. For correlation analysis, Shapiro-Wilk test for normality was used to determine whether data were normally distributed. Depending on normality, Pearson or Spearman correlation was used. Regression analysis was performed using lm() function. Regression assumptions were tested by graphic analysis. The influence of each data point on regression model was estimated with Cook’s distance. Significance level for all tests were α=0.05.
Results
Proliferation capacity and morphology of ADSCs during in vitro long-term cultivation
Growth kinetics
To assess cellular senescence in ADSCs, cell cultures from eight donors were subjected to long-term in vitro cultivation. For most donors, cells had been cryopreserved at P2 before beginning of the study (Table 1). After reaching confluency, a portion of cells was frozen for later analysis while the rest were reseeded to start the next passage. Subculturing was terminated if more than 4 weeks were necessary for cells to become confluent. Individual ADSC cultures reached the state of proliferation arrest after a considerably different time as were seen by differences in their respective cumulative PD values (Fig. 1A). Three cultures (CS-4, CS-5 and CS-7) stopped proliferating as early as after three (CS-4) or four (CS-5, CS-7) passages (cumulative PDs were 8.02, 8.30 and 10.19, respectively) and were excluded from further senescence evaluation as unsuccessfully expanded. In the remaining cultures, the number of cumulative PD varied from 14.69 (CS-6) to 28.97 (CS-8) (Table 1).
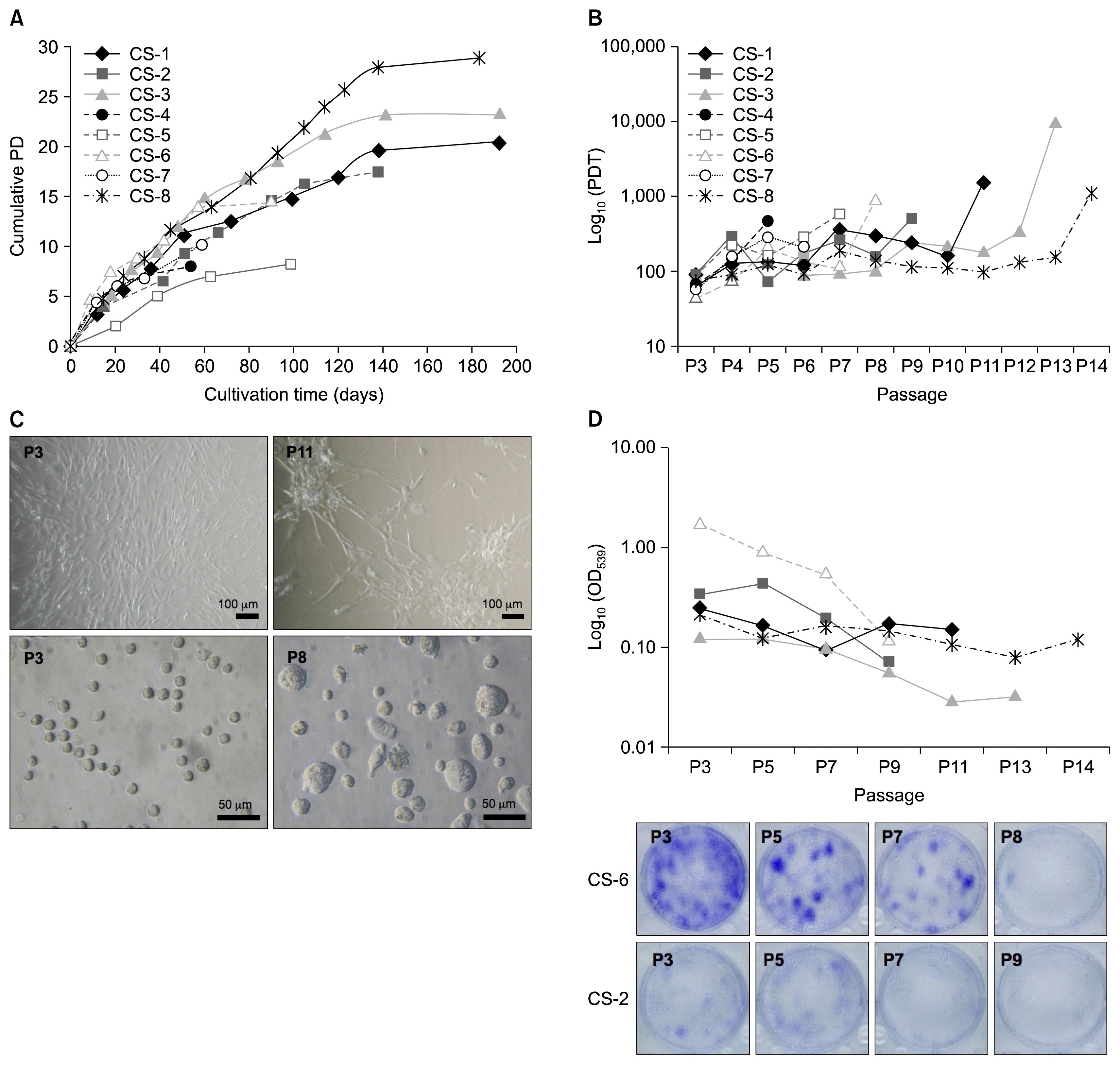
Fig. 1
Proliferation capacity and morphology of ADSCs during in vitro long-term cultivation. (A) Cumulative population doublings (PD). (B) ADSC proliferation curves showing population doubling time (PDT) at each passage. (C) ADSC morphology during long-term cultivation. Monolayer cells from early (upper-left) and late (upper-right) passage, and suspended cells from early (lower-left) and late (lower-right) passage are shown. (D) Clonogenicity potential of ADSCs. Upper panel –relative CFU test values; lower panel – an image of CFU test wells reflecting donor variance. OD: optical density.
![]()
The proliferation rates of all cultures decreased unevenly during expansion (Fig. 1B) despite maintaining consistent split ratio of equally dense monolayer cultures. One (CS-1, CS-3, CS-6, CS-8) or two (CS-2) pronounced peaks of increased PDT were observed in the middle part of cultivation followed by reactivation of proliferation in subsequent passages. Proliferative ability of all cultures was lost very rapidly at the last passage indicated by 3.7 to more than 10-fold increase of PDT comparing to penultimate passage. The arrested proliferation was also denoted by the minimal increase of cumulative PDs at the last passage (Fig. 1A). It has been reported that MSC proliferation potential lowers both with increasing time in culture and donor age (13). We found a positive regression between passage number and PDT (p<0.05) in samples CS-1 and CS-3, while in the case of CS-6 and CS-8, p value was close to significance level. Such relation was absent in CS-2 due to specifics of the growth curve. After the exclusion of unsuccessfully expanded cultures the rest of the samples fell into two distinct age categories – above 50 (CS-1, CS-2, CS-3) and under 40 years (CS-6, CS-8). There were no significant differences in growth kinetics between these groups, although the small sample size might compromise the validity of this observation.
Morphology
Morphologically, all cultures showed typical spindle-shaped appearance of MSCs at early passages. During long-term culture, the commonly described senescence-associated changes were observed: the increase of average cell size and heterogeneity, flattening of cells, irregular cell shape, and accumulation of granular inclusions in cytoplasm. Also, cells in late passages had a tendency to grow in star-shaped clusters rather than to form monolayers (Fig. 1C). However, similarly to previously described fluctuations in ADSC growth rates during culture, morphological changes did not accumulate evenly. Reappearance of spindle-shaped cells could be observed at P8 of sample CS-1 (Fig. S1). These cells dominated during P9 and P10 and possessed heightened proliferative abilities as indicated by decrease of PDT by 37.5% and 54.2% at P9 and P10, respectively when comparing to P7 (Fig. 1B). Similar observations were made in other ADSC cultures as well (data not shown).
At early passages, no significant differences in cell morphology were observed among donors. During long-term cultivation, donor-specific characteristics of cell appearance became visible (Fig. S2) indicating different course of aging in individual cultures. Nevertheless, all cultures reached terminal senescence having a typical look of aged cells and were unable to further divide.
Clonogenicity
Alterations in ADSC clonogenic property was assessed using CFU assay. 103 cells were seeded in a six-well plate in two replicates, cultured for 2 weeks and stained with crystal violet. Since we observed a tendency of cells to grow in a more diffused manner rather than form dense colonies the dye was dissolved after staining and quantified spectrophotometrically. Others have shown that colony forming ability negatively correlates with PDT, donor’s age and passage number (28). Our results showed a negative correlation between CFU test value and PDT, but it was not significant (p>0.05). Nevertheless, statistically significant negative relation between CFU test value and passage number existed (p<0.05). Again, the presence of donor variance was observed (Fig. 1D). This was illustrated by CFU test value of sample CS-6 which exceeded that of other cultures 7.47 times on average at P3. However, this difference rapidly declined during culture and reached the level of other samples at the last passage.
Senescence-related alterations of ADSC properties
Differentiation
Differences in adipo- and osteogenic potential between ADSCs from early (P3) and late (P9) passage were analyzed. Adipogenesis was assessed by staining intracellular lipid droplets with Oil Red O after 16 days of cultivation in adipogenesis induction medium. Osteogenesis was demonstrated by alkaline phosphatase activity and Alizarin Red S staining of accumulated calcium deposits after 30 days of differentiation. The results confirmed the ability of ADSCs to differentiate into the two lineages albeit with varying efficiencies among donors (Fig. 2). Accumulation of both lipid inclusions and extracellular calcium reduced dramatically at P9 compared to P3 (Fig. 2A, B). On the other hand, the level of alkaline phosphatase activity remained unchanged between passages (Fig. 2C).

Fig. 2
Bilineage potential of ADSCs. Shown is the comparison of early (P3) and late (P9) passage of two representative samples (CS-3 and CS-8) reflecting donor-specific differences. (A) Adipogenesis detected by Oil Red O staining of lipid inclusions. (B, C) Osteogenesis detected by (B) Alizarin Red S staining of extracellular calcium, and (C) alkaline phosphatase activity.
![]()
Senescence-associated β-galactosidase expression
As a common indicator for cellular senescence, expression of SA-β-gal was determined by a histochemical staining method. Every other passage of long-term culture-expanded ADSCs was analyzed. Images of non-overlapping fields of view were evaluated by two independent observers to quantify SA-β-gal expression levels. As expected, little or no SA-β-gal expression was detected in early passages with subsequent increase during further cultivation (Fig. 3A). This result was confirmed by regression analysis revealing a positive association between SA-β-gal expression value and passage number. However, this was not significant (p>0.05) due to fluctuations of SA-β-gal expression levels in samples CS-1, CS-3 and CS-8 (Fig. S3). Interestingly, these fluctuations were concomitant with those observed in growth analysis confirmed by statistically significant positive correlation (p> 0.05) between SA-β-gal expression level and PDT.
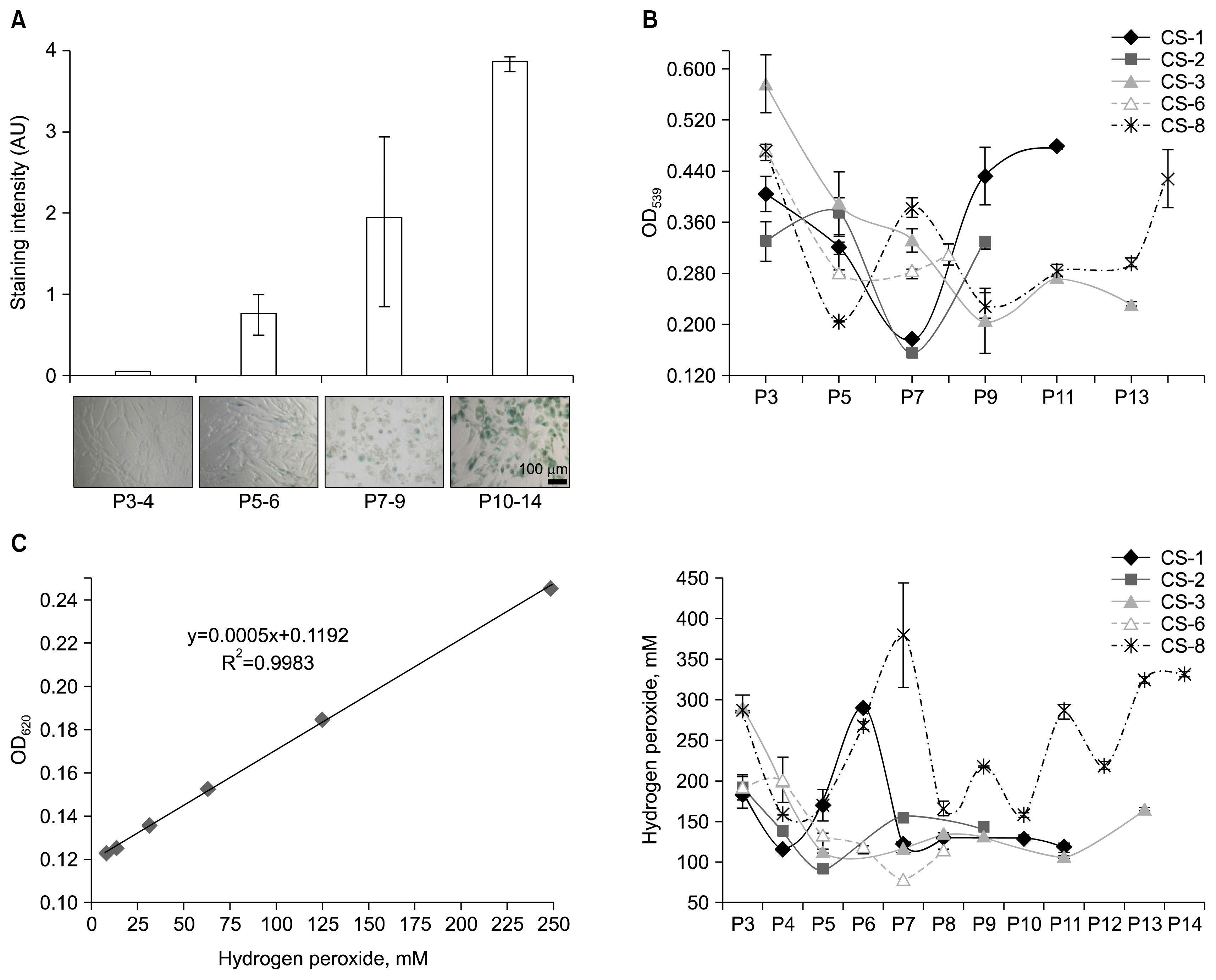
Fig. 3
Senescence-related changes of ADSC phenotype. (A) SA-β-gal expression was tested histochemically. Images were subjected to visual evaluation by two observers with regard to color intensity and percentage of stained cells per field of view. A typical view of increased SA-β-gal expression in ADSCs during long-term culture is shown (lower panel) with the respective visual evaluation data (upper panel) (B) ADSC metabolic activity determined by MTT assay. Optical density (OD) readings are expressed as mean of triplicate measurements. (C) Quantity of hydrogen peroxide in ADSCs. H2O2 standard curve is shown (left panel). H2O2 measurements of samples are expressed as mean of two replicates (right panel). AU: arbitrary units; P: passage. Error bars indicate standard deviation.
![]()
Metabolic activity
ADSC metabolic activity was assessed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Every other passage was tested. MTT test results showed no association with passage number in neither sample (p>0.05) indicating that, in our experimental conditions, changes of enzymatic activity in cells were not directly related to time in culture or PDT. However, the overall metabolic activity was higher in early passages, decreased during culture and increased once again towards the end of cultivation (Fig. 3B). This result might be explained by a shift in dominant cellular functions during long-term culture. Accordingly, elevated metabolic activity at early passages is consistent with rapid proliferation which ceases as cells approach senescence. In support of this, the lowest absorption values were detected at passages with proliferation slowdown peaks in growth curves (CS-1-P7, CS-2-P7, CS-3-P9, CS-6-P5) (Fig. 1B). The subsequent increase of metabolic activity at late passages could, on the other hand, be explained by such energy-consuming cellular phenomena as senescence-associated secretory phenotype (SASP), characterized by an active secretion of chemokines, cytokines, and other factors participating in inflammation, growth stimulation, tissue remodeling and matrix degradation, and autophagy, a process of nonspecific degradation of cytoplasmic components, which has been shown to be functionally linked to SASP, at least in certain cases (29). Thus, according to our data, increased MTT test values after a ‘fall’ might be associated with senescence.
Intracellular peroxide
Fluctuations of intracellular peroxide in ADSCs were measured quantitatively. Standard curve was generated using 2-fold serially diluted H2O2 starting with 1 mM standard. For precision, dilutions were prepared twice and each set of standards was measured in duplicate. Since the standard curve was not linear over the entire assay range only low end range was used (R2=0.9983) (Fig. 3C, left panel). The measurements of samples fell in this range.
Intracellular peroxide levels showed no association with neither passage number, nor PDT, nor metabolic activity (p>0.05). Peroxide concentrations varied among donors and there were substantial fluctuations even among passages of a single donor (Fig. 3C, right panel). Thus, no direct association between peroxide accumulation and senescence was proved in this study.
Gene expression profiles and transcription activities
To screen for alterations in gene expression in ADSCs during long-term culture 25 genes associated with such cellular functions as DNA repair, cell cycle regulation, antioxidation activity, apoptosis and epigenetic regulation were studied. Relative gene expression was determined by reverse transcription PCR and normalized against β-actin expression. The amplified products were visualized by ethidium bromide staining and quantified by densitometry analysis. Out of 25 genes tested, transcripts of three genes (TERT, BIRC5, GSTA5) were not detected in neither culture except CS-8 showing weak expression of BIRC5 (data not shown). For the rest of the genes, expression profiles were strongly donor-specific (Fig. S4) and rendered it impossible to associate the transcription of any of the genes with the development of senescence.
RTL and detection of ADSC subpopulations
To determine whether ADSC senescence phenotype was associated with alterations in telomere length, fluorescence in situ hybridization technique for flow cytometry (Flow FISH) was utilized using FITC-conjugated telomere-specific peptide nucleic acid (PNA) probes. The specific fluorescence was calculated by subtracting autofluorescence of cells without probe. Jurkat cells were added to each sample at a 1:1 ratio to normalize for inter-experimental variations. Cells in G0/G1 phase were discriminated by propidium iodide staining. Given the pseudodiploid karyotype of Jurkat cells (30) DNA index measurement was made. It was 1.00 for ADSCs (2n) and 1.83 for Jurkat cells (data not shown).
Results indicated that RTL significantly decreased in two samples (CS-3 and CS-8) when comparing the first and last passage (52.3% and 35.8%, respectively, p<0.05) (Fig. 4A). However, fluctuations of RTL were seen during passaging as in the previous tests indicating the complex nature of senescence process.
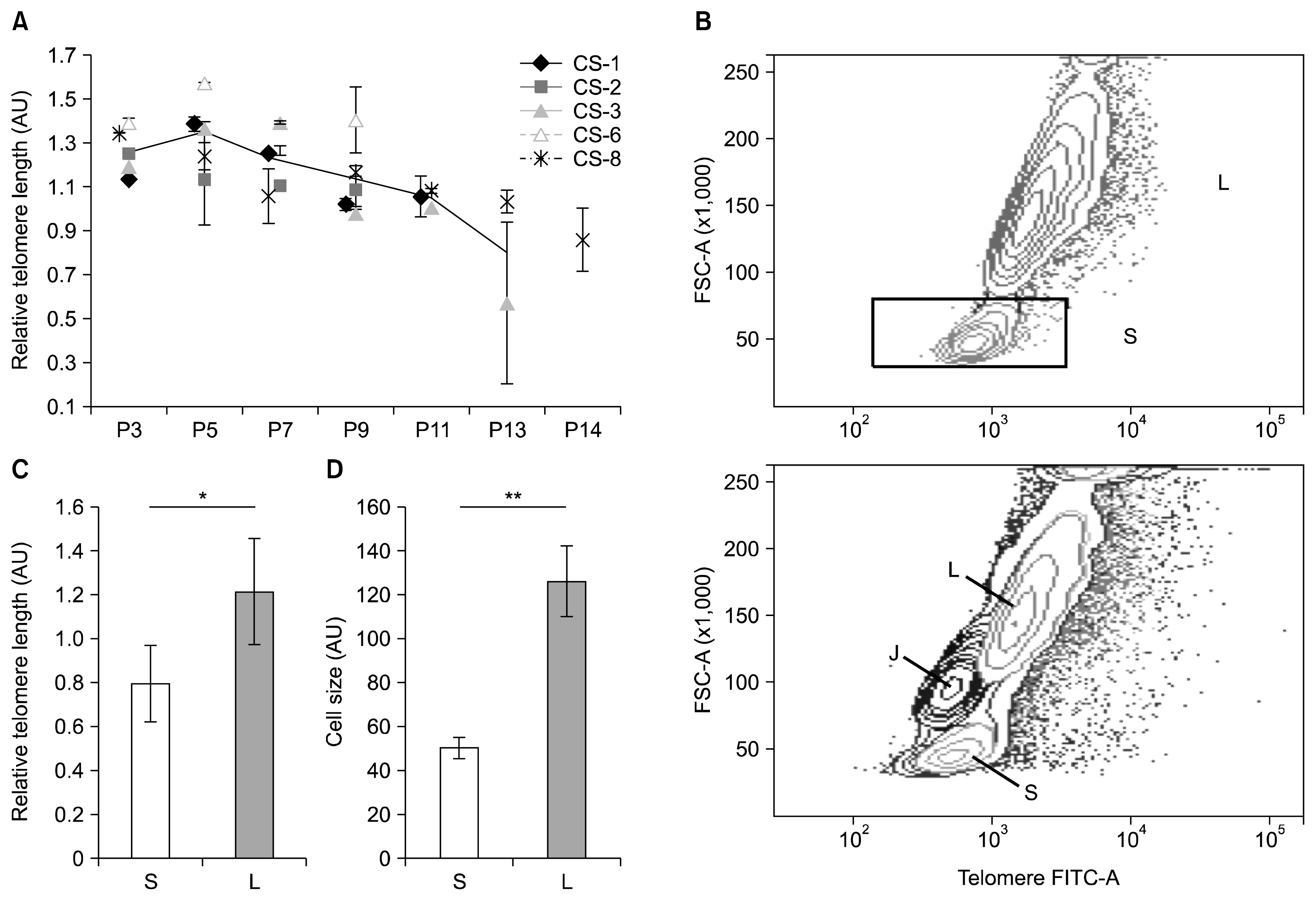
Fig. 4
Relative telomere length (RTL) of culture-expanded ADSCs and identification of cell subpopulations. (A) Flow FISH analysis of RTL dynamics during long-term culture. Each data point is expressed as mean±standard deviation of duplicate measurements. Continuous line represents mean RTL of five (P3–9), three (P11), and two (P13) samples. (B) The presence of two ADSC subpopulations (S and L) during RTL measurement was detected (upper panel). In contrast, only a single population of control Jurkat cells existed (lower panel, J). The S and L populations differed with respect to their RTL (C) and cell size (D) (data expressed as mean of all analyzed passages of five samples CS-1,2,3,6,8±standard deviation; *p<0.05, **p=0.001). AU: arbitrary units; P: passage.
![]()
To test whether the increase of telomere length observed in some passages could occur due to pseudo-lengthening of telomeres because of the presence of various cell sub-populations, forward scatter signal versus telomere-specific FITC fluorescence were analyzed. Surprisingly, two well-separated ADSC subpopulations were detected in all samples and passages analyzed albeit at varying quantitative relations among donors (Fig. 4B, Table 2). This result was specific to ADSCs since no such observation was made for simultaneously tested Jurkat cells (Fig. 4B, lower panel). Interestingly, the discovered subpopulations differed significantly in terms of RTL and cell size (p<0.05, Fig. 4C, D) and were designated as S population for ‘small’ cells and L population for ‘large’ cells. The mean RTL of S population was 0.95 to 1.75 times shorter than that of L population, while the difference of cell size was 2 to 3-fold depending on the sample.
Table 2
Comparison of relative telomere length (RTL), quantitative ratio and cell size of cell subpopulations S and L as identified by Flow FISH in adipose-derived mesenchymal stem cell cultures
![]()
As seen in Table 2, a gradual decrease of RTL during passaging was detected in both populations of CS-3 and CS-8 but for CS-1, only in L population (p<0.05). Furthermore, the shortening of RTL occurred more rapidly in L population compared to S in samples CS-1, CS-3 and CS-8, and, at the last passage, RTL difference between populations became insignificant. Since the cells lost their proliferative ability very rapidly at the last passage (Fig. 1A, B), this might indicate the critical telomere length blocking further proliferation. On the other hand, samples CS-2 and CS-6 followed a different pattern. For CS-6, there were no significant RTL changes during passaging in neither population (p>0.05), while for CS-2, changes were seen only in S population, but they were not significantly associated with passage number (p>0.05, Table 2). Thus, the senescence of these cultures might be caused by factors other than critically short telomeres.
During passaging, changes in quantitative relation between subpopulations were observed in a donor-specific manner (Table 2). When the dynamics of S population quantity and peaks of proliferation slowdown (Fig. 1B) were compared, it was found that peaks corresponded either to the highest percentage of S population cells (for CS-1, the amount of S cells was 2.6% in P3 and 10.9% in P7; for CS-8 – 3.0% in P3 and 32.6% in P7) or to substantial RTL decrease in the S population (for CS-3, RTL decreases by 35% in P9 compared to P7; for CS-2 – by 49% in P7 compared to P5).
Discussion
In this study, we aimed to analyze the development of senescence of eight ADSC cultures previously frozen and stored in liquid nitrogen for 1.5 to 3.5 years.
Originally, all cultures were obtained and cultivated at 5% oxygen in the presence of autologous serum before they were cryopreserved. By contrast, this work employed ambient oxygen and FBS as a serum source which might have impacted the course of senescence. However, such conditions mimic a realistic situation when patient’s cells have been cryopreserved in a tiered cell banking system, a recommended practice to minimize microbial contamination and facilitate large-scale cGMP-compliant cell production (31). In this case, cells can be stored in a master bank for an unknown period of time before further manufacturing is required. Due to evolving technological and regulatory requirements long-preserved cell products may become unintentionally exposed to suboptimal culture conditions.
MSC cultures are thought to be heterogeneous, comprised of subpopulations of cells with diverse features including multilineage potential, growth properties and clonogenicity (32, 33) During expansion, cell subsets possessing specific properties can be selected (34). Our data confirm the complex structure of MSC population by interrelated fluctuations of growth rate, morphology and SA-β -gal expression. Furthermore, an interesting observation was made by telomere Flow FISH when two distinct ADSC populations with significantly different cell size and RTL were found. The question remains whether this finding represents ‘real’ subpopulations of ADSCs. To prepare cells for RTL measurement they are subjected to harsh conditions such as high-temperature denaturation in the presence of a chaotropic agent (formamide) which is shown to have a detrimental effect on chromosomal morphology (35). Furthermore, Baerlocher and colleagues reported that heat and formamide treatment changed light scatter properties of human leukocyte subsets compared to untreated control (36). They also noticed swelling of cells upon prolonged exposure to hybridization mixture which depended on the composition of buffer used to suspend cells. Thus, forward scatter signals of S and L population are not necessarily an indication of the true size of these cells but should rather be perceived as a feature of tested ADSCs resulting in two distinct responses to treatment conditions as measured by Flow FISH. Nevertheless, the RTL difference of these two populations is clearly seen. Furthermore, unlike ADSCs, the control Jurkat cells, though undergoing the same treatment, retained their optical homogeneity and were not separated into distinct subpopulations, thus suggesting that the phenomenon is not a pure artifact.
Although several mechanisms of senescence are described, reduction of telomere length is studied in most detail. Other groups have reported mixed results concerning the role of telomere length in causing senescence (37, 38). The data obtained in this study allow us to conclude that both telomere-dependent and independent mechanisms could be observed in studied ADSC cultures. According to the telomere shortening pattern observed in S and L populations (Table 2) we hypothesize that three cultures (CS-1, CS-3, CS-8) entered replicative senescence (telomere length of L population reached that of – population at the end of cultivation), the other two (CS-2, CS-6) – premature senescence (telomere length of L populations did not change significantly). The three samples excluded from the study (CS-4, CS-5 and CS-7) are likely to be added to premature senescence group. It is reported that MSCs cease to proliferate when their telomeres reach approximately 10 kb in length (15). However, it is also known that critical telomere length can be as low as 4.5 kb (39). In our case, considering the reported average telomere length of Jurkat cells (11.5 kb) (40) that served as a control for RTL analysis, the extrapolated telomere lengths of L subpopulation at the last passage were 9.89, 6.78 and 8.28 kb in samples CS-1, CS-3 and CS-8, respectively which is in agreement with literature and the concept of replicative senescence. On the other hand, the same measurement revealed 13.57 kb in CS-2 and 17.82 kb in CS-6 further supporting the assumption of premature senescence in these samples. Moreover, the total number of PDs of these samples was lower than CS-1, CS-3 and CS-8 (Table 1) and the same result was seen in SA-β-gal test – the mean expression of the enzyme was 1.88 times lower at the last passage of CS-2 and CS-6 compared to the last passage of other samples (Fig. 4S).
According to our results, the preference for the two forms of senescence – replicative and premature – in the cells is dictated in a donor-specific manner. All ADSC cultures were initially obtained by a similar procedure, were all exposed to stress by changing culture conditions and propagated simultaneously under the same conditions. Nevertheless, one of the main observations in this study is the high level of donor variability. Diverse growth properties and functionality of MSCs obtained from different donors were observed previously (28) and are likely explained by different genetic backgrounds, clinical histories and lifestyle of individual donors.
Conclusions
The work presented here demonstrates that human ADSCs are heterogeneous and consist of cell subpopulations which play a major role in the course of in vitro senescence in every individual cell culture. Moreover, the pattern of in vitro aging is strongly donor-specific which my have important implications in personalized medicine.
 XML Download
XML Download