

 PDF
PDF Citation
Citation Print
Print


Introduction
EPCs are bone marrow-derived cells mobilized from the vascular niche to the peripheral circulation in response to tissue injury (1–5). Circulating EPCs home to damaged or ischemic microcirculatory beds where they actively participate in endothelial repair and neovascularization (6–8). Insulin resistance and hyperglycemia in patients with DM result in endothelial dysfunction by promoting the progression of atherosclerosis and cardio circulatory complications (9–12). It is well established that DM is associated to a reduced availability and downregulation of EPCs with an altered pattern of plasma growth factors and anti-oxidant gene expression (13–18). Interestingly, EPCs can be reduced in pre-diabetic states (impaired fasting glucose and impaired glucose tolerance) with further significant decrease at the time of clinical diagnosis of diabetes (19). The detrimental effects of DM on EPC number and function can impair vascular repair and regenerative activity and lead to vascular complications and worse clinical prognosis (20, 21). The definitive mechanisms responsible for the effects of hyperglycemia on EPCs needs to be elucidated. Decreased availability and down regulation of EPCs can occur as dysfunction in their mobilization, differentiation, migration and adhesion secondary to increased oxidative stress, reduced nitric oxide availability, altered secretion functions and derangement of intra-cellular signal pathways such mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinase (JNK) and phosphoinositide 3-kinase pathways (11, 22–26). When diabetic patients are compared to the group of control subjects, glycemic control exerts a positive influence on clinical incidence of ischemic events, cardiovascular accidents, and micro or macroangiopathies (18).
In this study, we analyzed the features of EPCs in severe Type 2 DM and the possible reversible effects of glycemic control. In particular, we evaluated whether a period of intensive glycemic control (three months) can influence the number and dynamics of mobilization of EPCs.
Materials and Methods
Type 2 DM patients and healthy controls between 50 and 85 years of age were eligible for inclusion in the study and enrolled from outpatient clinic basis. The overall risk factor load of an individual patient using a risk factor score including age above 40 years, male gender, hypertension, smoking, family history for cardiovascular heart diseases, and hypercholesterolemia, were calculated. Patients with type 2 DM (mean age 58.2±5.4 years, 25.6% women, disease duration of 15.4±6.3 years) had a baseline HgA1c of 8.7±0.5%. Exclusion criteria were the presence of acutely decompensate heart failure; a history of leucopenia, thrombocytopenia, or severe hepatic and renal dysfunction, evidence for inflammatory or malignant disease; or unwillingness to participate. Glicemic control was achieved in Type 2 diabetic group with a balanced dietary regimen, as previously described in detail (27). Diabetic patients were subjected to dietary evaluation (by food questionnaire) during the screening visit and monthly during the severe glucose control. Diabetic subjects were encouraged to control the body weight during the study and dietary regimen was provided. The proposed diet had a composition that was on average 27~28% fat (>80% in the form of monoinsatured fatty acids), 12~14% proteins and 52~58% carbohydrate (75% in the form of complex carbohydrate) while the colesterol intake was less than 210 mg day−1. Daily fibre assumption was greater than 58 mg. The study protocol was approved by the ethics review board and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from each patient.
Study protocol for isolation and count of EPCs
Mononuclear cells were isolated by density gradient centrifugation with Biocoll (Biochrom, Berlin, Germany) from 20 mL of venous blood as previously described in detail (4, 28). Briefly, after isolation, 4×106 mononuclear cells were placed on 24-well culture dishes blankets with human fibronectin (Sigma-Aldrich, Munich, Germany) and kept in endothelial basal medium (Cambrex, Walkerville, MD, USA) enriched with endothelial growth medium and 20% fetal calf serum. After the 4th day in culture, non-engrafted cells were detached by thorough washing with phosphate-buffered saline (PBS).
Characterization of late EPCs
Was assessed the uptake of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-labeled acetylated low-density lipoprotein (DiLDL) by cells as described in detail (4, 28). Incubation of cells with DiLDL (2.4 μg/mL) was performed at 37°C for 1 hour. Then, cells were fixed with 2% paraformaldehyde for 10 min and incubated with fluorescein-5-isothiocyanate (FITC)-labeled agglutinin I (lectin, 10 μg/mL; Sigma-Aldrich, Milan, Italy) for 1 hour. Dual-staining cells positive for both lectin and DiLDL were considered as EPCs. The endothelial features were additionally documented by flow cytometry analysis of vascular endothelial growth factor receptor 2 (KDR) and von Willebrand factor (4, 28). The number of EPCs was evaluated by counting carefully selected high-power fields (4, 28).
Measurement of functional capacity of EPCs
Isolated EPCs were separated using 1 mmol/L ethyl-enediaminetetraacetic acid in PBS (pH 7.4), harvested by centrifugation, resuspended in 500 μL endothelial basal medium, counted, and placed in the upper chamber of a modified Boyden chamber (2×104 cells; BD Bioscience, Milan, Italy) as described in our previous studies (4, 28). For identification, cell nuclei were marked with 4′,6′-diamidino-2-phenylindole. Cells migrating into the lower chamber were quantified in several random microscopic fields (4, 28).
Bone marrow mononuclear cells
Bone marrow-derived mononuclear cells (BM-MNCs) were isolated from bone marrow aspirates by density gradient centrifugation. After two washing procedures, we performed a cell suspension using an X-vivo medium (Cambrex, Walkerville, MD, USA) with heterogeneous cell populations including hematopoietic progenitor cells (4, 28).
Flow cytometry analysis of BM-MNCs
For the identification of hematopoietic stem/progenitor cell populations cells were added with conjugated antibodies against human CD45 (mouse FITC-labeled; BD Pharmingen, Milan, Italy), human CD34 (FITC-labeled and allophycocyanin-labeled; BD Pharmingen) and human CD133 (allophycocyanin-labeled; Miltenyi Biotec, Bologna, Italy) (4, 28).
Colony-forming unit assay
The BM-MNCs (1×105 per well) were founded in methylcellulose plates (Methocult GF H4535, Stem Cell Technologies, Milan, Italy) enriched with stem cell factor, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, IL-3, and IL-6. Plates were analyzed under phase-contrast microscopy, and granulocyte-macrophage colony-forming units (CFU-GM, colonies >50 cells) were counted at the 14th day of incubation (4, 28).
Measurement of High-sensitivity C-reactive protein levels
High-sensitivity C-reactive protein from serum of type 2 DM patients and healthy controls was measured by means of particle-enhanced immunonephelometry (Dade Behring, Marburg, Germany) as previously described (4).
Statistical analyses
The non-parametric Mann-Whitney U test was used to assess differences between two groups. Categorical variables were compared by the chi-square test or the Fisher exact test. Bivariate correlation was calculated by Pearson correlation. A linear regression model was used to evaluate independent predictors. If not stated otherwise, data are expressed as mean±SD. Statistical significance was assumed at p≤0.05. All statistical analyses were performed using SPSS for Windows version 12.0 (SPSS Inc., Chicago, IL, USA).
Results
The characteristics of the study population are described in Table 1. The two populations (healthy controls and Type 2 DM patients at baseline) were comparable for age, gender, for smoking activity and for NYHA functional class. Significantly differences were assessed for number of hypertensive subjects, family history for CHD, for LDL cholesterol, and for use of drugs as statins, ACE-I/ARB, beta-blockers and diuretics in the DM group. Finally, High-sensitivity C-reactive protein was higher in Type 2 DM patients at baseline compared to healthy control group.
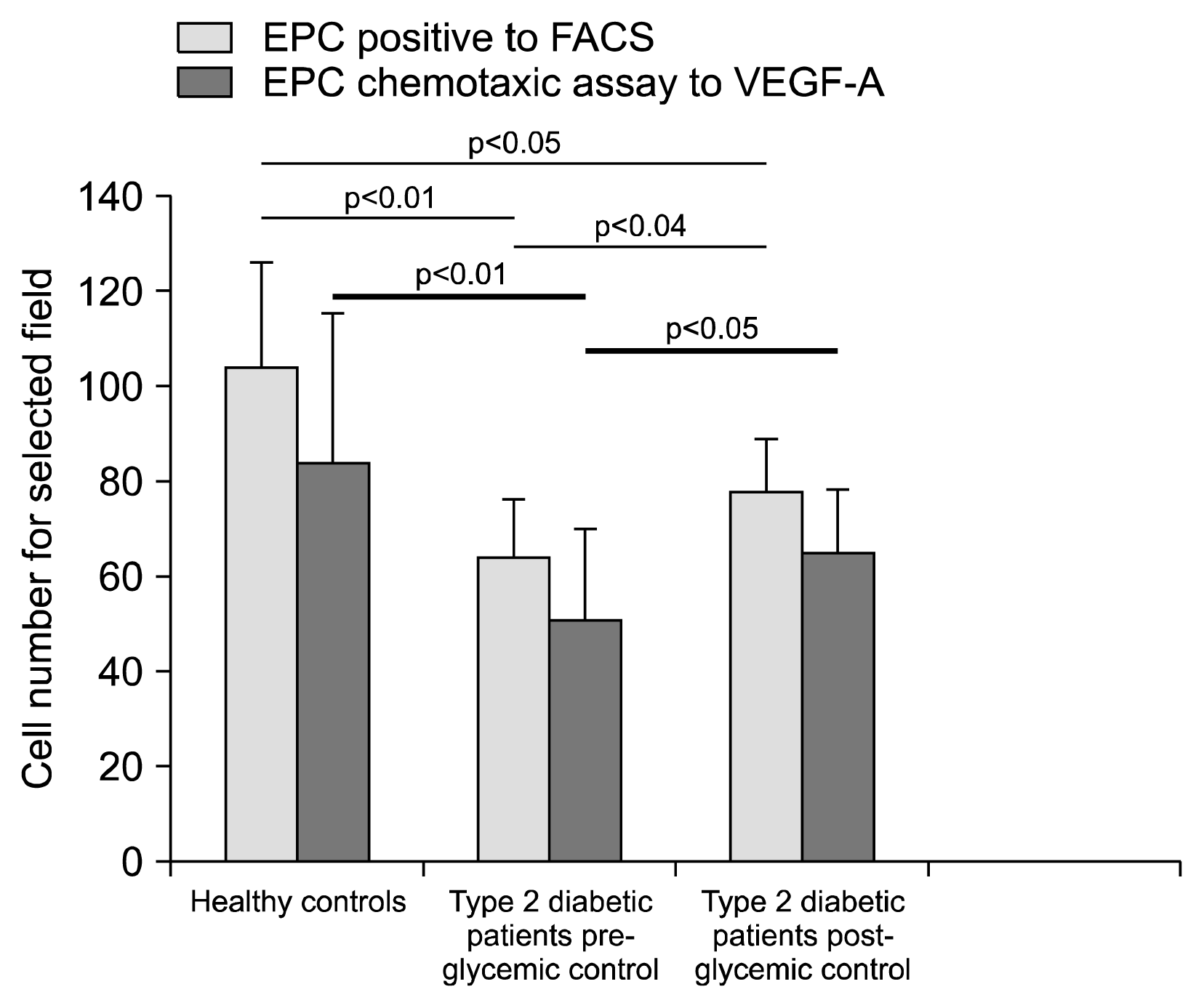
As shown in Table 2, in Type 2 DM baseline, the main determinants of EPC cell number on univariate analysis were advanced age, elevated serum levels of hs-CRP and the status of diabetes. When we considered the multivariate analysis, only the presence of Type 2 DM was an independent predictor of reduced numbers of circulating EPCs, whereas all other parameters lost predictive power (Table 2). Thus, in our experimental conditions, Type 2 DM itself was an independent determinant of circulating EPC levels. The numbers of hematopoietic progenitor cells, by which the circulating EPCs presumably rise, were determined by the expression of the marker protein CD34 by flow cytometry analysis. EPC levels (CD34+/KDR+) were lower in Type 2 DM patients in comparison to healthy controls (64±13 vs 104±21, p<0.01). Relevantly, the number of EPCs in patients with Type 2 DM increased after three months of glycemic control (64±13 baseline vs 78±11 post glycemic control, p<0.04) (Fig. 1). However, the difference in EPC level (CD34+/KDR+) (FACS analysis) between the healthy controls and Type 2 DM patients, also after the glycemic control, remained statistically significant (p<0.05) (Fig. 1).
Functional capacity of circulating EPCs at baseline and after glycemic control
The functional capacity of circulating EPCs was assessed by measuring their migratory attitude in response to VEGF. EPCs derived from patients with Type 2 DM showed a significant impaired migratory capacity assessed by EPC chemotaxis assay toward VEGF-A compared with EPCs derived from healthy controls (Fig. 1, p<0.01 vs controls). After glycemic control, migratory capacity significantly improved compared to pre-glycemic control (p<0.05) but was still significantly lower than in healthy controls. In Type 2 DM patients, on univariate analysis, the main determinants of EPC functional capacity at baseline were advanced age, elevated serum levels of hs-CRP, the family history for CHD and the status of diabetes (Table 3). After glycemic control, the only determinant was the elevated levels of hs-CRP. However, on multivariate analysis, only the presence of Type 2 DM was an independent predictor of reduced migratory capacity of EPCs (Table 3). Taken together, Type 2 DM is associated with a reduced number of circulating and functional impairment of EPCs.
Functional capacity of BM-MNCs
The expertise of progenitor cells in the BM aspirates was determined by measuring CFU-GM activity. The BM-MNCs derived from Type 2 DM patients showed a significantly reduced number of CFU-GM compared to BM-MNCs from healthy controls (51±19 vs 84±32 number of colonies per 105 cells, p<0.02 vs controls). The number of EPC CFUs also increased significantly after glycemic control (65±14 number of colonies per 105 cells, p<0.05 vs baseline). In Type 2 DM patients at baseline, on univariate analysis, the main determinants of CFU-GM were advanced age, sum risk factors, number of CD34+CD45+ and CD34+CD133+ cells (Table 4). However, on multivariate analysis, only the presence of Type 2 DM was an independent predictor of reduced numbers of CFU-GM at baseline and after glycemic control.
Discussion
This study confirms that EPC number and bioactivity are reduced in patients with Type 2 DM and, most importantly, shows that an intensive glycemic control in the same group of patients can restore EPC number with less pronounced but still significant improvement in their function. There is a wide evidence on the key role of circulating EPCs in the maintenance of endothelial integrity, function and angiogenesis (29, 30), with growing interest on their use as a novel therapy for cardiovascular diseases (2, 7, 31). DM is associated to decreased EPC level and function leading to a rapid development of atherosclerosis, microangiopathies and cardiovascular accidents (13–19, 25, 26, 32, 33). EPCs can be considered biological markers in the DM patients and prognostic indicators of future cardiovascular complications in these subjects (33–37). Exhaustion of the pool of progenitor cells in the bone marrow, the impaired functional capacity within the bone marrow, the reduced mobilization, survival and/or differentiation of EPCs could account for the lower circulating EPC number in DM patients (2, 7, 37). Previous studies, showed that DM patients with good glycemic control have a higher circulating EPC number and a better function compared to DM patients with inadequate glycemic control (18, 19, 32). Our data demonstrated that a relatively short period of intensive glycemic control in Type 2 DM patients can reverse the tendency to decline of EPC level and positively affect EPC function. We showed a close relationship among glycemic control, number and migratory response of EPCs in these patients. We adopted a standardized protocol, which showed a high capacity to detect specifically EPC subpopulations (late EPCs) (4, 28). However, we advocate that further standardization of laboratory techniques for the detection of EPCs as FACS protocols could better compare different data concerning several studies. Here, we confirm and extend previous observation that DM patients show a functional impairment of EPCs both in the BM as well as in peripheral blood as circulating progenitors. A relevant data was that, after adjusting for other cardiovascular risk factors and medications, the glycemic control was identified as an independent variable for EPC count and function. On the other hand, the circulating EPC number and bioactivity in DM patients after glycemic control did not reach the EPC level of healthy controls as observed by previous studies (32). Interestingly, in type 2 DM patients, the glycemic control has a greater impact on the number of circulating EPC with respect to the increase of CFU. This data suggests that EPC migratory response is less restored in comparison to EPCs levels. We believe that these results need to be confirmed in larger cohorts of diabetic subjects considering also extended periods of glycemic restriction. Our results indicate that in Type 2 DM patients, glucose control may be considered a mechanism to preserve EPC pool inverting the diabetic degenerative pattern coupled with endothelial dysfunction, progression of atherosclerosis and cardio circulatory complications.
This study is only an observational research. Additional investigation is needed to explore the mechanism of the increase of EPCs after glucose control. Furthermore, taking into consideration the different treatment categories used by enrolled patients, we suggest randomized-controlled clinical trials focusing on the effect of single anti-diabetic drugs on EPC pool (16, 38, 39). Further researches are needed to validate our findings in patients subjected to different pharmacological treatments in addition to the dietary regimen.
 XML Download
XML Download