

 PDF
PDF Citation
Citation Print
Print


Introduction
Hematopoietic stem and progenitor cells (HSPCs) possess the capacity for self-renewal (mitotic division leading to production of the same stem cells without loss of developmental potential) and differentiation (asymmetric division leading to unequal production of daughter cells from the same mother cells). Though these two opposing forces of self-renewal and differentiation act on HSPCs, hematopoietic homeostasis is maintained throughout the lifetime of an individual by carefully orchestrated sequence specific transcription factor networks, genes and signaling pathways (1, 2). Transcription factors recruit cofactors to gene regulatory regions, which facilitate or inhibit gene transcription by chromatin modification (3). Modulation of gene expression by chromatin modification on global and gene-specific level is ‘Epigenetic’ regulation. Since the hematopoietic cell lineage development follows a strict hierarchical pattern emanating from a primary single HSC, the hematopoietic system provides an ideal model system to study the correlation between epigenetically directed changes in chromatin structure and gradual restricted cell potential during differentiation. DNA organized in loose chromatin (euchromatin) is readily available for gene expression and contribute for the maintenance of pluripotency. Conversely, DNA tightly packed into dense chromatin (heterochromatin) has genetic code buried effectively and become inconspicuous for genetic reading and transcription. Epigenetic reprogramming describes stable and inheritable alterations in gene expression that occur without permanent changes in DNA sequence. Such changes include active or repressive histone modifications working in closed concert with DNA methylation (2). Major covalent modifications that occur at histone tails are acetylation/deacetylation by histone acetyltransferases/histone deacetylases and methylation/demethylation by histone methyltransferases/histone demethylases. Methylated histone arginine and lysine residues are considered static modifications because of low levels of methylgroup turnover in chromatin. With the identification of enzymes that counteract histone methylation, dynamic nature of these modifications has emerged (Fig. 1) (4). In contrast to a phenomenal variety of histone modifications, DNA methylation is simply binding of a methyl group at carbon-5 position of cytosine base by DNA methyltransferases (DNMTs), primarily in cytosine-phosphate-guanine (CpG) dinucleotides or CpG islands (5). DNA methylation leads to stable long-term repression. While histone methylation events may be associated with gene activation, certain forms of histone methylation cause a local formation of heterochromatin, which is readily reversible. Apparently, DNA methylation and histone modifications are dependent on one another, their cross-talk is mediated by biochemical interactions between SET histone methyltransferases and DNA methyltransferases (6). The demethylation process is quite complex and involves proteins, such as the TET (ten-eleven translocation) family of proteins. TET1 was found to catalyze the oxidation of 5-methylcytosine, a well-known epigenetic mark, into 5-hydroxymethylcytosine in mammalian DNA (7). Interestingly, regulation of promoter demethylation-associated 463 genes is more frequent in the hematopoietic system than in non-hematopoietic tissues (241 genes), supporting the overall influence of demethylation signals in driving the tight time-and step-controlled hematopoietic cell fate determination. For example, promoter methylation of hematopoietic genes is linked to stemness as demethylation occur during early embryonic development and later on during hematopoietic differentiation. Genome wide methylation status of induced pluripotent stem cells (iPSCs) and somatic CD34+ cells further confirm the role of DNA methylation in the regulation of hematopoietic gene expression. DNA methylation functions during iPS cell generation by silencing of genes important in differentiation, and facilitating chromatin remodelling. Conversely, DNA demethylation appears to play an important role in reactivating pluripotency genes, particularly in the late stages of the reprogramming process. Repressive epigenetic modifications, (H3K9me3 and DNA methylation) at the promoter regions, occasionally observed in partially reprogrammed iPS cells, fails to produce the robust reactivation of pluripotency genes (8). The acetylation of histones form the relaxed chromatin, conducive to gene expression activation. The primary site of histone acetylation is the addition of acetyl moiety to the ε-amino terminal of lysine residues, which change the physicochemical properties of histone proteins, interfering with the electrostatic attraction between positively charged histones and negatively charged DNA. In contrast, the removal of acetyl groups from the acetylated histones, “histone deacetylation”, forms the basis of inactive chromatin environment (heterochromatin) (2). Stem cells in the primitive and differentiated stage organize their level by multitude layers of molecular epigenetic events to impose flexible but precise control to establish important regulatory gene expression patterns. In this review, we highlight recent findings in epigenetic regulation in hematopoietic stem cells and their progeny. We also delineated the mechanism of aberrant placement of epigenetic marks and mutations in the epigenetic machinery involved in hematopoietic malignancies.
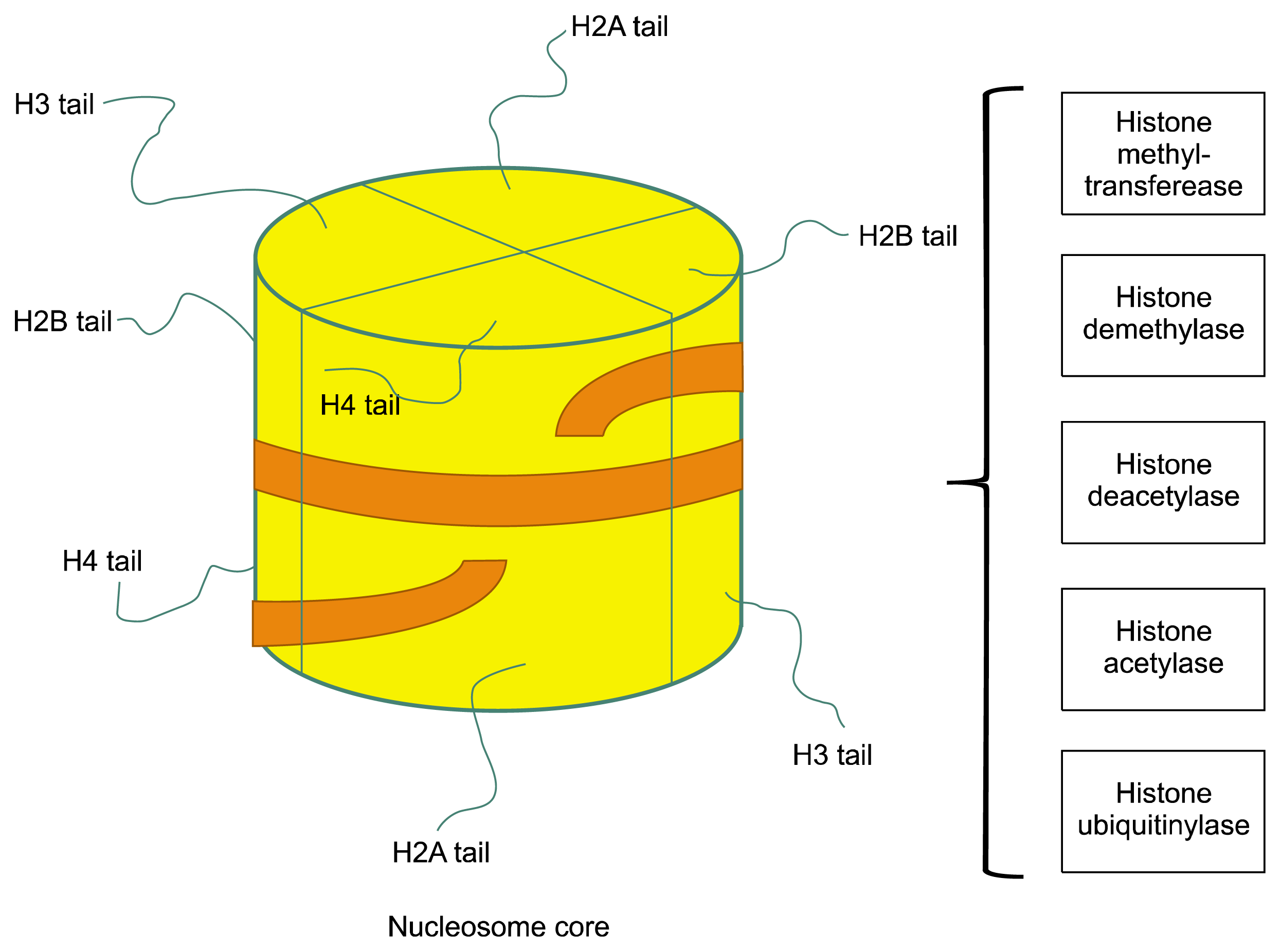 | Fig. 1Schematic of nucleosome structure. Nucleosomes are the smallest structural unit of chromatin. Nucleosome consists of two copies of each core histone (H2A, H2B, H3 and H4) and ~150 bp DNA. The N-terminal tail of each histone is extruded from the nucleosome. Amino acids in histone tails can be modified by numerous enzymes bringing acetylation, methylation, phosphorylation, ubiquitination and other substitutions, creating a complex ‘bar’ code, which may influence chromatin structure by affecting histone-histone and histone-DNA interactions. Post-translational modifications of histones regulate transcription of genes important for self-renewal and differentiation.
|
Go to : 
Epigenetic events from self-renewal to differentiation
DNA methylation
Stem/progenitor cell differentiation is linked to gradual methylation primarily in cytosine-phosphate-guanine (CpG) dinucleotides or CpG islands that reside within DNA regulatory sequence by DNA methyltransferases. However, whether DNA methylation induces transcriptional silencing or preserves silence state of chromatin, remains enigmatic (2). DNMT3a and DNMT3b show complementary de novo methylation patterns responsible for silencing of self-renewal genes in HSCs (9). Consistent with this, loss of DNMT3a results in expansion of HSC population by impeding differentiation and upregulation of self-renewal genes (Runx1 and Gata3). Combined loss of DNMT3a and DNMT3b in HSCs result in enhancement of HSC self-renewal by activating β-catenin signalling (10). Methylation is maintained by DNMT1 to permit efficient hematopoietic differentiation. DNMT1 is crucial for the progression of stem cells to multipotent progenitors to lineage-restricted myeloid progenitors and regulating cell cycle entry (11). DNA methylation is known to interplay with other chromatin marks, such as histone modifications (12). Extrinsic factors also effect epigenetic regulation. The dynamic interplay between the epigenetic changes and gender-specific hormone apolipoprotein E (Apoe) provide insights for the modulation of a reconstituting potential of HSPCs. In a study, female mice transplanted with truncated DNA methyltransferase 3B isoform, DNMT3B7, resulted in very high expression levels of Apoe. The CpGisland controlling Apoe expression had lower levels of modified cytosines in DNMT3B7 transgenic HSPCs. DNMT3B7 expression down-regulate hematopoietic number within the female hormonal microenvironment (13). Stem cell protein, SALL4, recruit DNMTs to silence genesto govern-stem cell self-renewal. It has been investigated that DNMTs and histone deacetylase repressors interact synergistically to reverse the transcriptional repression effect of SALL4 (14). Overexpression of SALL4 leads to increased methylation of silenced genes (H3K4me3 and H3K79me2) in primary HSPCs. During normal hematopoiesis and leukemogenesis, SALL4 mediated expression up-regulates multiple regulatory genes including HOXB4, Notch1, Runx1, Meis1 and Nf-ya, influencing particularly, three important self-renewal pathways involving Bmi1, β-catenin and PTEN. As SALL4 regulate apoptotic pathways both in normal HSCs and leukemic stem cells, Gao et al., deciphered that targeting SALL4 combined with BCL2 antagonist (ABT-737), could lead to leukemic stem cell-specific apoptosis (15). The precise combination of genes which on activation/repression control the processes of driving proliferation and suppressing differentiation, have yet to be defined.
Some genes mutated in cancer recruit histone modifying enzymes and thus alter gene expression. However, the radix of the aberrant methylation of target genes in the tumorous cell is not fully elucidated. Truncated DNMT3B proteins are expressed in primary acute leukemias (2). The PML-RAR gene translocation and RUNX1 in acute promyelocyticleukemia recruits DNMTs to target promoters that change the active chromatin structure to silence status and contributes to its leukemic transformation (16). Recently, hypomethylating agents emerged as a standard for treatment in myelodysplastic syndrome as they reprogram “methylome” and re-establish hematopoiesis (17).
Histone acetylation
Protein acetylation regulates HSC self-renewal, proliferation, and their differentiation into committed hematopoietic progenitors. Histone acetyltransferases (HATs) acetylate histone proteins by transferring the acetyl group from acetyl-CoA to specific lysine residues, resulting in a dispersed structure of chromatin, accessible for transcriptional factors. HATs contain p300/CBP (CBP and p300), MYST (Tip60, MOZ, MORF, HBO1 and HMOF), and GNAT (PCAF, Gnc5 and ELP3) family members to regulate normal and malignant hematopoiesis. Multiple chromatin factor complexes, such as NuA4/P300/CBP/HBO1 are required for normal hematopoiesis (17). CBP/p300 regulates self-renewal and differentiation in adult HSCs through regulating Gfi1b, monocyticleukemia zinc finger protein (MOZ) maintains the generation and development of HSCs and promote proliferation of progenitors by repressing the transcription of p16. It has been proposed that the fusion proteins produced upon translocation of the human MOZ locus with CBP/p300, support the development of leukemia by altering the regulation of MOZ transcriptional targets (18).
Histone deacetylase inhibitors result in chromatin remodeling to modulate re-expression of silenced tumor suppressor genes in leukemic stem and progenitor cells which, in turn, result in cellular differentiation, inhibition of their proliferation and self-renewal properties (19). Histone deacetylase inhibitor, valproic acid (VPA) increases self-renewal of HSCs by blocking differentiation at multiple regulatory pathways. Epigenetically reprogrammed CD34+ cells on treatment with VPA generate greater numbers of functional stem cells and multilineage hematopoiesis with capacity for bone marrow reconstitution before clinical transplantation. Mechanistically, VPA progress to cell cycle by down-regulating p21, activating Hoxb4 and wnt-signaling pathway (20). Walasek et al. showed synergistic effects of VPA and lithium to induce self-renewal by blocking differentiation genes and preserve expression of stem cell-related genes (21). VPA inhibits proliferation and induces apoptosis in leukemic cells by increasing expression of death receptors and their ligands in acute promyelocyticleukemia and AML-ETO dependent leukemia. Aberrant formation of fusion proteins through chromosomal translocations of HAT and HAT-related genes (e.g. MOZ, MORF, CBP and p300) occurs in leukemia. Mistargeting of such deleterious fusion proteins contributes to global alterations in histone acetylation patterns in cancer. Specific epigenetic gene silencing can occur by aberrant targeting of histone deacetylases to the gene promoter which causes histone hypoacetylation (22).
Histone methylation
Epigenetic cross-talk between DNA methylation by DNA methyltransferases and histone methylation by histone methyltransferases have ramifications for the understanding of gene transcription for normal development and aberrant gene silencing in tumors. It is not yet clear whether DNA methylation patterns guide histone modifications (histone acetylation and methylation) during gene silencing/or DNA methylation take its cues primarily from histone modification status (23). Histone methylation is a reversible process: methyl marks are established by histone methyltransferases, so-called “writers,” and removed by histone demethylating enzymes, termed “eraser”. Three methylation sites on histones are implicated in activation of transcription: H3K4, H3K36, and H3K79. Two of these, H3K4me and H3K36me, have been implicated in transcriptional elongation. Three lysine methylation sites are connected to transcriptional repression: H3K9, H3K27, and H4K20. Methylation at H3K9 is implicated in the silencing of euchromatic genes as well as forming silent heterochromatin. Like lysine methylation, arginine methylation correlates with both permissive and non-permissive chromatin states. Enzymes (protein arginine methyltransferases, PRMT’s) are recruited to promoters by transcription factors (24). Open chromatin and transcriptionally active regions are most commonly recognized by two different methylation status of H3K4, H3K4 dimethylation (H3K4me2) and tri-methylation (H3K4me3). H3-K4me3 is a key hallmark of active promoters. H3K4me2 marks both promoters and enhancers, and commonly occur at CpG islands. Genes associated with silenced DNA and heterochromatin are generally associated with methylations at lysine 9 or 27 of histone H3 (H3K9 and H3K27). Coexistence of repressing H3K27me3 and activating H3-K4me3 epigenetic marks on same DNA region, usually at regulatory regions of gene promoters, are called ‘bivalent domains’. This bivalency fine-tunes gene expression during development and contemporarily, keep gene expression in a poised or primed state that allows for either rapid activation or stable silencing upon differentiation of hematopoietic cells (Fig. 2) (25).
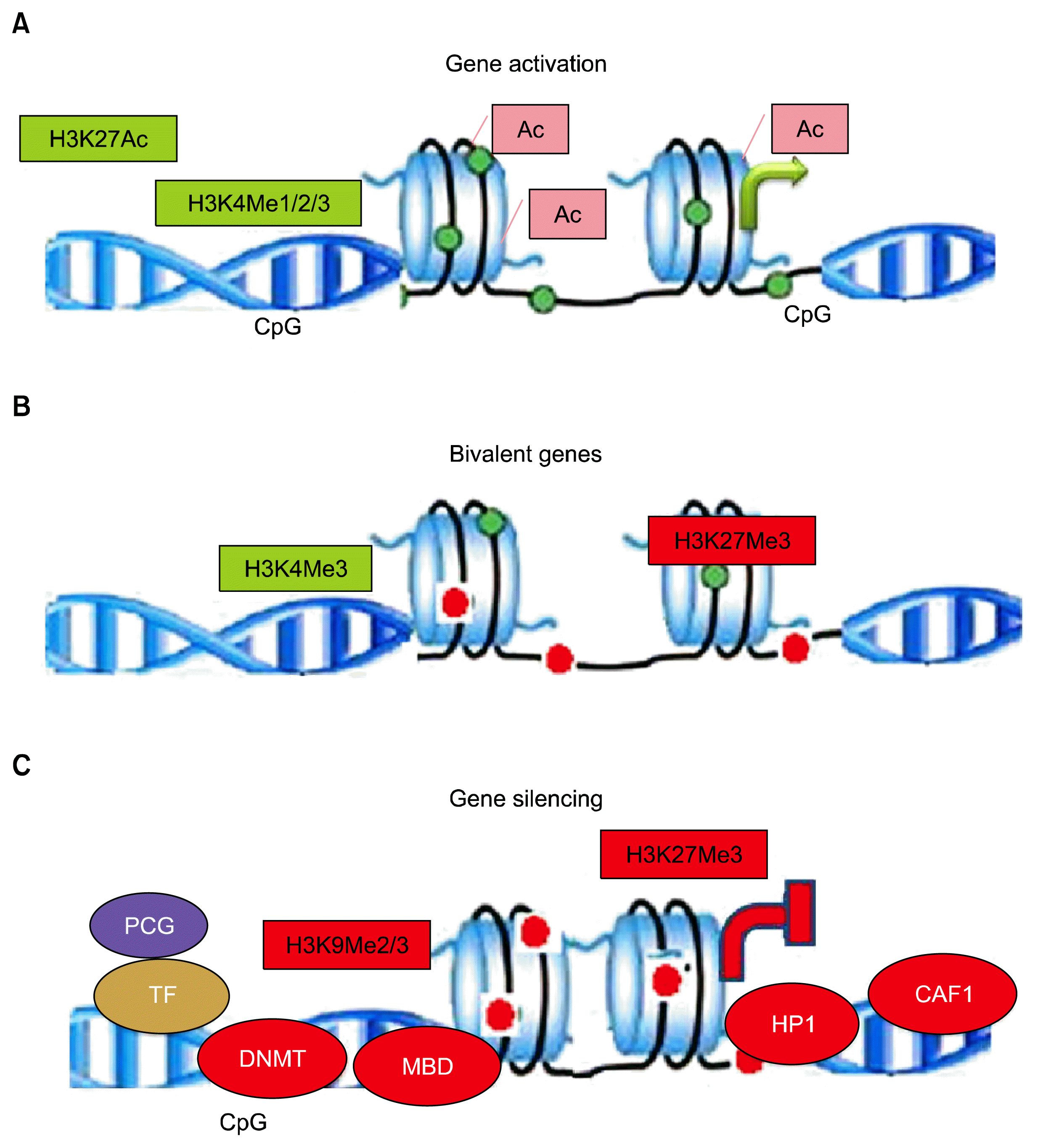 | Fig. 2Histone modifications: Active genes: Open chromatin structure of transcriptionally active gene with loosely spaced nucleosomes. Acetylation of lysine neutralizes the positive charge, reducing affinity between histone and DNA, which functions as platforms for the recruitment of transcription factors or chromatin remodelers, thus histone modifications directly effects nucleosomal architecture. H3K4me3 is enriched around transcription start sites. H3K4me1 is enriched around enhancers and downstream. H3K27ac is enriched around active enhancers and transcription start sites. Bivalent genes: In undifferentiated stem cells, both H3K4me3 and H3K27me3 (active and inactive marks, respectively) are enriched around transcription start sites on many genes. The multiple coexisting histone modifications are associated with activation and repression. Inactive genes: H3K9me3 is broadly distributed on inactive regions. H3K27me3 and H3K9me3 are usually not colocalized. Proteins associated for transcription silencing are DNMT- DNA methyl-transferase, MBD-Methyl-binding domain, HP-1-Heterochromatin protein and CAF-1-Chromatin assembly factor1.
|
Chromatin-modifying polycomb group (PcG) proteins form large multimeric structures distinctly, polycomb repressive complex 1 and 2 (PRC1 and PRC2). The PRC1 includes BMI1, RING1/2, MEL-18, RAE28/MPH1, and M33/CBX2. PRC1 represses genes by mechanisms such as chromatin compaction and decreasing nucleosomal turnover. As observed in knockout mouse lines for Ring1B, Bmi1, Rae28, and Mel18, mice harboring mutations in the components of PRC1 generate hematopoietic abnormalities. Once circumscribed to the chromatin, Ring1B and Bmi1 ubiquitinate histone H2A at lysine 119 (H2AK119ub) (26), which is considered to be involved in transcriptional repression by inhibiting transcription initiation or by restraining RNA polymerase II from elongation. It is not yet confirmed that whether PRC1 function as a transcriptional repressor is dependent or independent of its H2Aub ligase catalytic activity (27). PRC2 stabilize repressive chromatin structure through the function of chromatin modifiers such as enhancer of zeste (EZh2), embryonic ectoderm development (Eed) protein and suppressor of zeste 12 (Suz12). PRC2 is responsible for di and tri-methylation of lysine 27 of histone H3 (H3K27me2/me3) to promote transcriptional repression. HSCs express high levels of Bmi1, a member of the PcG of transcription repressors. Furthermore, PRC1 component Bmi1 plays an important role in HSC self-renewal and lymphocyte differentiation by transcriptional repression of genes involved in cellular senescence (p16Ink4a, p19Arf, and p53) (28). Increased expression of the p53 target gene, Wig1 in Bmi1−/− bone marrow suggests that p19Arf pathway gets activated in Bmi1deficient hematopoietic cells (29). Bmi1 is critical in preventing Ikaros-mediated lymphoid priming, thus, prevents premature lineage specification of HSPCs (30). It has recently become clear that Bmi1 also functions in the protection against oxidative stress. Bmi1 is frequently overexpressed in human hematopoietic malignancies. Down modulation of Bmi1 impairs self-renewal and long-term expansion of leukemic stem cells. Bmi1 also functions in an orchestration of the symmetry of HSC divisions and bypassing senescence (31). Changes in the expression of individual PcG genes cause perturbations in the composition of the PRC, which affect overall PRC enzymatic activity. Overexpression of BMI1 is associated with malignant hematopoiesis (32). EZH2 over-expression direct stem cells toward a cancer stem cell state. EZH2 serve as a tumor-suppressor gene as its inhibition in cancer stem cells offers a potential therapeutic approach to counter the aberrant activation in lymphoma and leukemia (33).
Histone demethylation
Histone demethylases (HDMs) reportedly reshape the epigenome of hematopoietic stem cell and thus regulate its fate. There are two families of HDMs, the amine oxidase enzyme, known as lysine demethylase (LSD1) and the Jumonji C (JmjC) domain-containing family. LSD1 forms a core complex with corepressors, including HDAC1/2 and CoREST and repress the transcription of target genes. LSD1 specifically removes one or two methyl groups from histone H3 lysine 4 (H3K4) or 9 (H3K9), depending on the cellular context and the presence of cofactors, thereby repressing or activating transcription. Overexpression of the shortest isoform of LSD1, which is specifically repressed in quiescent HSCs, demethylates histone H3K9 more efficiently than other isoforms (34, 35). LSD1 mediates erythroid differentiation via epigenetic modification of the GATA-2 locus and also increase self-renewal potential via upregulation of the HoxA family but retains differentiation ability towards T-cell lineages at transcriptome levels in HSCs. LSD1 has been identified as component of a transcription activation complex containing MLL1. The dysregulation of MLL in acute myeloid leukemia suggests that LSD1 may also have links to leukemogenesis. LSD1 overexpression seemingly is directly implicated for priming of HSCs towards malignant transformation and progression to T-cell leukemogenesis. Thus, provides a novel strategy for early diagnosis and effective treatment of the disease (36).
The second family of HDMs includes the JmjC enzymes, which catalyse the removal of mono-, di- and trimethyllysines. Several JmjC domain proteins have been identified with different specificities including JHDM1, JHDM2, JMJD2 and Jumonji AT-rich interactive domain 1 (JARID1). The interaction of the JmjC HDM JARID1d with the polycomb-like protein RING6a/MBLR showed that RING6a/MBLR enhance JARID1d-mediated H3K4 demethylation, associated with transcriptional repression. JHDM1b has also been shown to interact with PcG proteins in a complex containing Ring1b/Rnf2 and the Bcl6 interacting co-repressor among other proteins (37). This co-repressor complex may be implicated in gene repression mediated by Bcl6, a transcription factor frequently dysregulated in B-cell lymphomas. Deletion of JARID1b compromises HSC self-renewal capacity, which suggests that JARID1b is a positive regulator of HSC potential (38). JARID1b/KDM5b is highly expressed in primitive hematopoietic compartments and is overexpressed in acute myeloid leukemias (5).
Histone ubiquitination
Among the histone octamer, H2A is the most abundant ubiquitinated protein and PRC1 is the main H2A ubiquitin (H2Aub) ligase. Ubiquitination of histone H2A (H2A-K119u) is a common chromatin modification associated with gene silencing, and controlled by the activities of ubiquitin ligating and deubiquitinating enzymes. The RING1 subunit of PRC1 monoubiquitinylates histone H2A at lysine 119 (H2A-K119u), which promote transcrition repression and chromatin compaction. Another H2A-deubiquitinating enzyme, Mysm1 plays a critical role in HSC maintenance, self-renewal, genetic stability in hematopoietic progenitors, and development of lymphoid and erythroid lineages (39, 40). Despite its important function and early discovery, histone ubiquitination remains the least understood compared with other histone modifications.
Go to :
Conclusions
The cumulative research data demonstrate the crucial role of epigenetic players in stem cell self-renewal and differentiation gene expression patterns. The startling development of reprogramming technology has catapulted stem cell role in regenerative medicine and disease modeling and offers new hope for personalized regenerative cell therapies. Recently developed high-throughput genomic tools greatly facilitate the study of epigenetic regulation in pluripotent stem cells by mapping chromatin-nuclear matrix interactions. Elucidating epigenetic landscape will have important implications to expedite manipulation of stem and progenitor cells for regenerative applications and would render novel targets to tackle human diseases prospectively, leading to new therapeutic avenues in regenerative medicine.
Go to :
 XML Download
XML Download