

 PDF
PDF Citation
Citation Print
Print


Introduction
Oxygen (O2), a critical factor for the survival of all aerobic organisms, is the final electron acceptor in mitochondrial oxidative respiration, and impairment of oxygen availability leads to the loss of cellular function or even cell death. Although 21% of ambient oxygen is used in the conventional stem cell culture protocol, stem cells including embryonic and adult stem cells in the body have available a relatively low concentration of oxygen at 2 ~ 9% due to physiological reasons such as vascularization (1). In this respect, distinguishing the functional differences arising from the oxygen concentration is important in understanding the nature of stem cells and in controlling stem cell fate for therapeutic purposes. Reactive oxygen species (ROS) are more reactive than free oxygen and are well known for their critical roles in the regulation of the developmental processes, such as the emergence of embryonic blood stem cells or differentiation of embryonic cardiomyocytes (2, 3). ROS are also involved in many biological processes including gene transcription, protein translation, and protein-protein interactions (4). ROS may function as cellular signaling molecules involving the propagation of signaling and translation of environmental changes into cellular responses in order to maintain cellular homeostasis. There is also increasing evidence about the various ways in which ROS coordinate various cellular processes and adapt cellular activity to bioenergetic sources (5). These previous reports implied that the changes in oxygen level and ROS might act as mediators for the communication between the mitochondria and the nucleus (6, 7). Recent findings strongly indicate that ROS have important roles in both stem cell maintenance and their differentiation, which provide the possibility that the regulation of ROS in stem cells could be a valuable tool for developing stem cell-based therapeutics. This review article attempts to discuss the physiological role of ROS in stem cell fate decision and its effect on metabolic regulation of stem cell.
Physiologically relevant oxygen level for stem cells
Recent advances in physiological relevance between oxygen and stem cells have revealed that oxygen acts as an important signaling molecule as well as a critical factor in the stem cell niche. Although an oxygen concentration below atmospheric (containing 21% of oxygen) is considered as hypoxia, most types of cells among mammalian cells have available only approximately 2~9% of oxygen, with this concentration of oxygen actually representing physiological normoxia (1). Tissue oxygen concentration is depended on the blood supply, and the blood oxygen concentration drops to about 5~10% in venous blood (8). Vascular distribution and a decline in the oxygen concentration of the blood make a difference in various tissues: bone marrow, 1~7% (9, 10); brain, 0.5~8% (11, 12); mammary gland, 1.7~6.8% (13), and adipose tissue, 3 ~ 5% (14). The preimplanted embryo lacking a vascular system encounters a low oxygen concentration of 2.3%, which rises up to 8% after implantation with access to maternal vasculature (15). Thus, the term hypoxia denoting an oxygen level lower than the 21% of the atmosphere is revealed to be really a physiologically normoxic condition for the cells. For this reason, to express actual tissue normoxia, some authors suggest a new word ‘physioxic’ rather than hypoxia. In traditional in vitro cultivation conditions developed in cells with high growth rates such as fibroblasts or cancerous cell lines, cells are cultured with 5% CO2, with approximately 21% atmospheric oxygen. This culture condition is suitable for certain cell lines, but stem cells require a more specific microenvironment reflecting their in vivo niche.
It is still controversial whether the low oxygen concentration supports stem cell maintenance and differentiation in vitro. However, the impact of physiological oxygen concentrations on embryonic stem cells and hematopoietic stem cell cultures is well established. Unlike adult stem cells, ESCs propagated for many passages in an in vitro condition of 20~21% O2 are well adapted to a high oxygen concentration and result in a higher dependency on oxidative phosphorylation than that of glycolysis. Despite the adaptation to higher oxygen levels, these cells maintain a relationship between stemness and hypoxic condition. In general, hypoxic condition is needed for the pluripotency of ESCs. With conventional culture conditions at higher oxygen levels, ESCs spontaneously lose undifferentiation marker gene expression such as OCT4 and SSEA4 and differentiate into other types of cells (16, 17). In contrast, a hypoxic culture condition with an oxygen level below 5% supports the maintenance of ESC pluripotency and control embryonic stem cell development, which is mediated by hypoxia inducible factors (HIFs) (1, 18–20). Furthermore, oxygen gradients act as guidance for placenta, trachea, and cardiovascular system development, which imply that low oxygen levels are required to control embryonic development (1).
Regulation of ROS production and deletion in stem cell
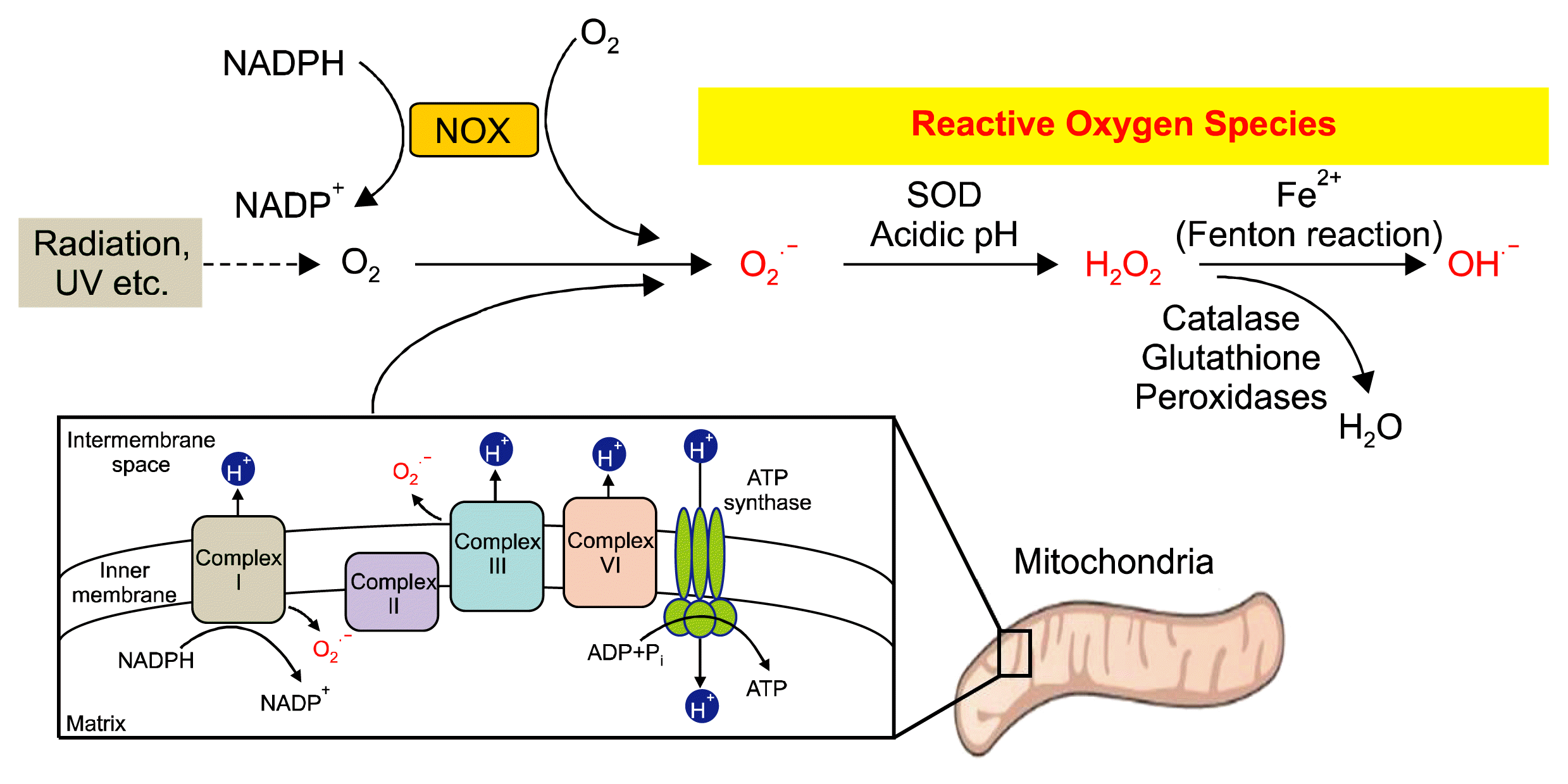
A high concentration of oxygen in the atmosphere and its oxidative nature enables it to produce oxidized biological macromolecules and results in the generation of reactive intermediates known as reactive oxygen species (ROS). ROS, chemically reactive molecules, are generated by the one-electron reduction of the oxygen molecule, a type of radical anion. There are three different forms of intracellular ROS: superoxide anions (O2−), hydrogen peroxide (H2O2) and hydroxyl radicals (OH−). Historically, ROS were considered harmful byproducts that escaped during the metabolic process, but accumulating evidence has shown that ROS have an important role as a signaling mediator in cell fate decision (21, 22). Among ROS isoforms, H2O2 is known as the most potent ROS involved in intracellular signaling and acts as second messenger, integrating and delivering environmental stimuli to the downstream signal cascade. This is due mostly to the rapid reduction of superoxide anion to H2O2 by superoxide dismutase (SOD) (23), as well as its longer half-life and membrane permeability (Fig. 1) (4).
ROS are produced by conserved biochemical reactions in response to the cellular environment, which can largely be divided into intra- and extra-mitochondrial processes. During the generation of the proton motive force for ATP production by electron transport chain, approximately 0.1~0.2% of O2 consumed by the mitochondria is converted to ROS, which occurs mainly through complexes I and III of the electron transport chain (24). Complex I elicits the proton motive force by passing the electrons through the membrane-bound enzymes of redox centers, such as flavin mononucleotide (FMN) and 8-iron-sulfur (FeS) clusters, produced by the oxidation of nicotinamide adenine dinucleotide (NADH) (25). Complex III also contributes to superoxide generation, which passes electrons from ubiquinol to cytochrome C (26). Although their contributions are less well characterized, Complex II is also involved in mitochondrial ROS generation. Although complex II mutations can generate ROS, the enzyme has limited capacity to produce superoxide compared to complex I or III due to the suppression of flavin radical production (27). Complex IV also has catalytic activity to reduce O2 to H2O, but this complex does not contribute to mitochondrial ROS generation. Extra-mitochondrial ROS generation is associated with the membrane-bound protein NADPH oxidase (NOX), which produces O2− and, subsequently, H2O2 with NADP+ (28). In addition to NOX, xanthine oxidase, uncoupled endothelial NO synthase, cytochrome p450, heme oxygenase, peroxisomes, myeloperoxidase, and lipoxygenase (28) are also involved in extra-mitochondrial ROS generation. Although the contributions are not characterized, metabolic enzymes and intermediate metabolites such as dihydroorotate dehydrogenase (29), α-ketoglutarate (30), and pyruvate dehydrogenases (31) are also associated with ROS generation (25).
To protect against oxidative damage from excessive accumulation of ROS, ROS removal is finely controlled through the ROS scavenging system, which maintains the redox balance of cells. Cells and body fluids contain antioxidants that neutralize or scavenge ROS. Antioxidants including superoxide dismutase (SOD), catalase, peroxiredoxins (PRX), thioredoxin (TRX), glutathione peroxidase (GPX) and glutathione reductase (GR) can directly react with ROS and take away the electrons. Among the antioxidants, glutathione (GSH), one of the most abundant and potent antioxidants in the cell, reduces oxidized proteins and H2O2 through the glutaredoxin and thioredoxin system. In addition, SOD and catalase are also involved in ROS removal through the reduction of O2− and H2O2, respectively. Cellular redox homeostasis controlled by ROS production versus antioxidant defense is critical for the regulation of both physiological and pathophysiological cellular functions.
Role of ROS on stem cell fate
ROS can regulate the nutrient-sensing pathway through direct interaction with metabolic enzymes and proteins (32, 33), which could affect cellular processes such as cell cycle progression, apoptosis, quiescence, or differentiation (34, 35). The previous evidence suggests that ROS act as signaling mediators linking between metabolic alteration and stem cell fate. Therefore, the elucidation of ROS regulation on stem cell proliferation and maintenance of self-renewal capacity will provide new insight into the optimization of in vitro stem cell culture systems, embryonic development, and the regulation of stem cell fate for therapeutic applications of stem cells. Although excessive ROS accumulation induces oxidative damages including apoptosis, ESCs possess the ability to resist oxidative stress (36). Consistently, under the physiological normoxic condition (2%) of their niche, the genomic integrity and pluripotency of ESCs remain intact (37), but prolonged hypoxia exposure results in elevated intracellular ROS levels which subsequently induce apoptosis (38). Furthermore, hypoxia-induced alteration of metabolic flux such as an elevated dependency on glycolysis and the pentose phosphate pathway rather than oxidative phosphorylation contributes to maintaining ESC self-renewal by shortening the G1 cell cycle phase (39–41). Rapid ATP generation and increased nucleotide biosynthesis through glycolysis and the pentose phosphate pathway, respectively, are required for rapid proliferation and DNA synthesis (42). Similarly, previous reports have shown that the inhibition of uncoupling protein 2 (UCP2)-induced oxidative phosphorylation deteriorates stem cell properties and lead to differentiation or apoptosis (41). However, metabolic shift from oxidative phosphorylation to glycolysis through HIF-1α activation under hypoxic condition stimulates stem cell proliferation and maintenance while suppressing differentiation (43, 44). In mouse ESCs, SIRT1, which elevates intracellular ROS levels by inhibiting p53 antioxidant function, is involved in the expression of pluripotency marker (45). In addition, it has been reported that fork-head box protein O1 (FoxO1) is associated in maintaining human ESC pluripotency (46) through interaction with transcription factors involving pluripotency regulation, such as OCT4 and NANOG. FoxO1 also promotes cell cycle arrest and induces apoptosis by modulating the genes involved in DNA repair and oxidative stress defense (47). Furthermore, microRNAs are also involved in ROS-mediated stem cell fate such as self-renewal and differentiation (48). Upregulation of miR10b and miR23b make MSC more susceptible to oxidative stress through inhibition of p38 MAPK or antioxidants genes such as TXNL2 and GPx3 (49). In contrast, miR210 alleviate oxidative stress through c-Met pathway repression (50). These results indicate that pluripotency is maintained through a highly complex pathway, and it can be altered by manipulating the metabolic pathways through genetic approaches and drugs as well as the intracellular ROS levels.
Despite much effort to develop the optimal differentiation process, the current differentiation of pluripotent stem cells or induced pluripotent stem cells has depended on using factors or exogenous gene insertion associated with embryogenesis. In this respect, ROS could be an alternative factor in the regulation of stem cell differentiation, and the elucidation of the roles of ROS and their mechanism is needed. In the uterus, the pre-implantation embryo is faced with a relatively hypoxic environment, and during initial implantation, to meet the increased demands for oxygen and substrates, vasculogenesis/angiogenesis occurs, which is coincident with a metabolic shift from glycolysis to oxidative phosphorylation and a reflection of an evolutionarily optimized process (51). Similar to ESCs, elevated intracellular ROS levels in adult stem cells are required for differentiation into specific lineages, including osteogenesis (52, 53), suggesting a critical role for ROS in the stem cell differentiation process (54). Physiologically, ROS could accumulate by the impairment of redox homeostasis, such as an increase in ROS generation and/or decrease in antioxidant defenses. The capacity to resist the oxidative damages of ESCs and MSCs correlates with the constitutive expression of antioxidant enzymes. In previous reports showing the role of NOX in the differentiation of ESCs, high glucose in the culture medium increased the mitochondrial ROS level and subsequently activated p38 MAPK through NOX4, which stimulated differentiation toward the cardiomyocyte lineage (55, 56). These results suggest that ROS induced by high glucose are needed for efficient differentiation of ESCs toward cardiomyocytes (57). Similarly, the involvement of NOX4 in mouse ESC differentiation toward smooth muscle cell (SMC) lineage has also been reported (58). Transforming growth factor β1 (TGF-β1)-mediated NOX4 activation promotes SMC differentiation by generating H2O2, while inhibition of NOX4 significantly decreased intracellular ROS levels in differentiating ESCs. ROS also affected adipocyte differentiation of MSCs by acting as an upstream regulator of CREB which induces C/EBPβ expression and subsequently elicits adipocyte differentiation of MSCs. These ROS-induced differentiations of MSCs towards adipocytes were blocked by inhibiting NOX4 (59, 60). The differentiation process is closely associated with mitochondrial biogenesis and maturation to fulfill the energy demand required for specialized functions during lineage-specific differentiation (39). To meet the energetic demands, the dependency of mitochondrial oxidative phosphorylation increases which subsequently increases intracellular ROS levels and could contribute to defining cellular fate. According to stem cell differentiation, mitochondrial maturation (revealed as morphological changes from spherical into tubular structures), mtDNA replication, and expression of key enzymes involved in the tricarboxylic acid cycle (TCA) and oxidative phosphorylation are accelerated to support the increasing mitochondrial functions (40, 61–64). In addition, the alteration of metabolic phenotypes to oxidative phosphorylation according to elevated ROS levels from differentiation through the suppression of the ROS scavenging system and downregulation of NADPH generation, also suggest the role of ROS as a mediator between metabolism and differentiation.
Metabolic regulation of ROS in stem cells
Effect of ROS on glycolytic metabolism in stem cells
Embryonic stem cells (ESCs), derived from the inner cell mass (ICM) of blastocysts, divide rapidly while maintaining their pluripotency (65). In addition, ESCs have immature mitochondria indicating reduced mitochondrial function, which mainly is due to low a membrane potential, low content of mtDNA, spherical morphology, and sparse density with predominantly perinuclear localization of the mitochondria (40, 52, 63, 64, 66, 67). Consistent with an immature mitochondrial structure, ESCs have low energy turnover and ATP production compared to differentiated cells (40, 64, 66, 68). Thus, to meet their energetic demands and alleviate ROS-dependent DNA damage, ESCs have distinct mechanisms such as preferring glycolysis, elevated antioxidant defense, prevention of DNA damage, and apoptosis. ESCs largely depend on glycolysis rather than mitochondrial oxidative phosphorylation in glycolytic metabolism, in contrast to their differentiated counterparts (66, 69). Increasing metabolic flux through glycolysis and the pentose phosphate pathway enhances NADPH generation, which acts as a crucial cofactor for supporting the scavenging of ROS by maintaining thioredoxin and glutathione. The protection mechanisms from ROS through glycolysis-dependent metabolism are well known in mouse embryonic fibroblasts, in which the over-expression of glycolytic enzymes such as phosphoglycerate mutase (PGM) and glucose-6-phosphate isomerase (GPI) increases glycolysis and reduces oxidative damages (70). Consistently, blockage of the pentose phosphate pathway by a glucose-6-phosphate dehydrogenase knock-out makes ESCs more susceptible to oxidative stress (71). Their utilization of glycolysis over mitochondrial oxidative phosphorylation may thus have a role in the protective metabolic mechanism and as an alternative energy source to meet the demands of pluripotent stem cells.
Although pluripotent stem cells can grow and survive in a wide range of oxygen concentrations from 3% to 20%, limiting oxygen availability under the circumstances of a preimplanted and implantated embryo requires a metabolic shift to anaerobic glycolysis to produce sufficient ATP for embryo development and cellular homeostasis. This hypothesis is supported by the fact that hypoxic condition reflecting their niche environment stimulates glycolysis (72) and intensifies the maintenance of stemness and the self-renewal capacity (12, 16, 73). In addition, the hypoxic environment also can elevate the efficiency of nuclear reprogramming to produce induced-pluripotent stem cells (iPSCs) (74). As complementary protection against ROS-induced damages through a nonoxidative metabolic phenotype, pluripotent stem cells possesses a strong ROS scavenging system than that of their differentiated counterparts, which is involved in the reduction of intracellular ROS through high expression of antioxidant enzymes including thioredoxin-glutathione reductase, glutathione peroxidases, glutathione-S-transferase, and superoxide dismutase 2 (SOD2). In addition, stress response mechanisms working through verapamil-sensitive multidrug efflux pump, heat shock protein expression, and DNA strand- break repair processes enhance the physiological protective capacity against oxidative damages (75, 76). Similar to pluripotent stem cells with distinct differences in glycolytic metabolism and ROS generation, iPSCs derived from nuclear reprogramming also have an immature mitochondria structure and a reduction in mtDNA (40, 52), as well as transcriptional regulation of nuclear genes involved in oxidative phosphorylation and glycolysis. Indeed, these metabolic differences could be anticipated based on the induction of pluripotent markers and the efficiency of iPSC production, suggesting that the dependence on glycolysis may also affect the progression of nuclear reprogramming (66, 77).
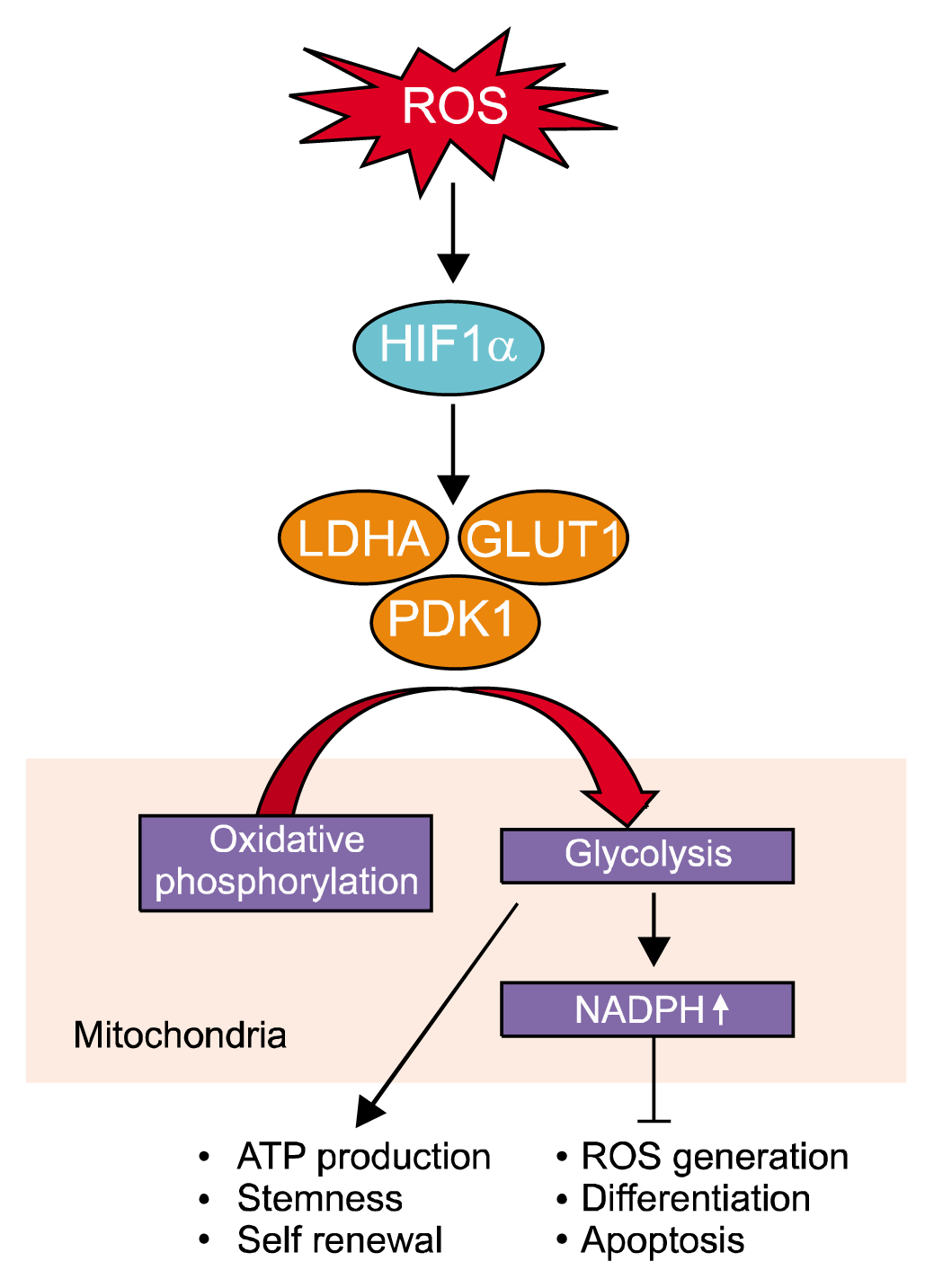
Adult stem cells have a limited cell-cycle progression and are maintained in a quiescent state within a niche of differentiated tissue where the O2 is maintained at 1~8% (78). Among the niches of adult stem cells in various tissues, the hematopoietic stem cell niche is best characterized. This niche is comprised of the bone marrow microenvironment, and stromal and progenitor cells, which can affect the oxygen gradient within the common space and compete for oxygen (79). Under hypoxic condition, quiescent hematopoietic stem cells resist damage accumulation from oxidative stress and maintain a more robust ability to repopulate. Hypoxia-induced inhibition of ubiquitination and proteasomal degradation results in the accumulation hypoxia-inducible factor-1α (HIF-1α), enabling the binding and activation of hypoxia response elements in target genes (79). HIF-1α, which is highly expressed in hematopoietic stem cells, should be regulated within a narrow range. In HIF-1α inhibition, the HIF-1α pathway deteriorates hematopoiesis making cells susceptible to stress resistance, and subsequently eliciting embryonic lethality or cellular apoptosis; however, excessive expression or accumulation HIF-1α simulates destabilization and premature exhaustion of hematopoietic stem cells. HIF-1α promotes metabolic shift from oxidative phosphorylation to glycolysis by regulating gene transcription involved in glycolytic metabolism, including glucose transporter 1 (GLUT1), lactate dehydrogenase A (LDHA), and pyruvate dehydrogenase kinase (PDK1) (Fig. 2) (79). In addition, Meis1-mediated increase of HIF-1α transcription is also involved in the maintenance of hematopoietic stem cells metabolic phenotypes such as glycolysis dependency and the potential suppression of ROS production (80, 81). Similar with hematopoietic stem cell niche, neural stem cell niche is under hypoxic condition, which is required for the maintenance of the undifferentiated state (82). Taken together, hypoxic signaling has a critical role in the maintenance and glycolytic metabolism regulation of adult stem cells in their niche, which suggest the possibility that the regulation of glycolytic metabolism through ROS could be applicable to various types of stem cells because hypoxia extends lifespan by increasing proliferation capacity and reduces differentiation of additional stem cell populations (12, 16, 74, 83).
Effect of ROS on amino acid metabolism in stem cells
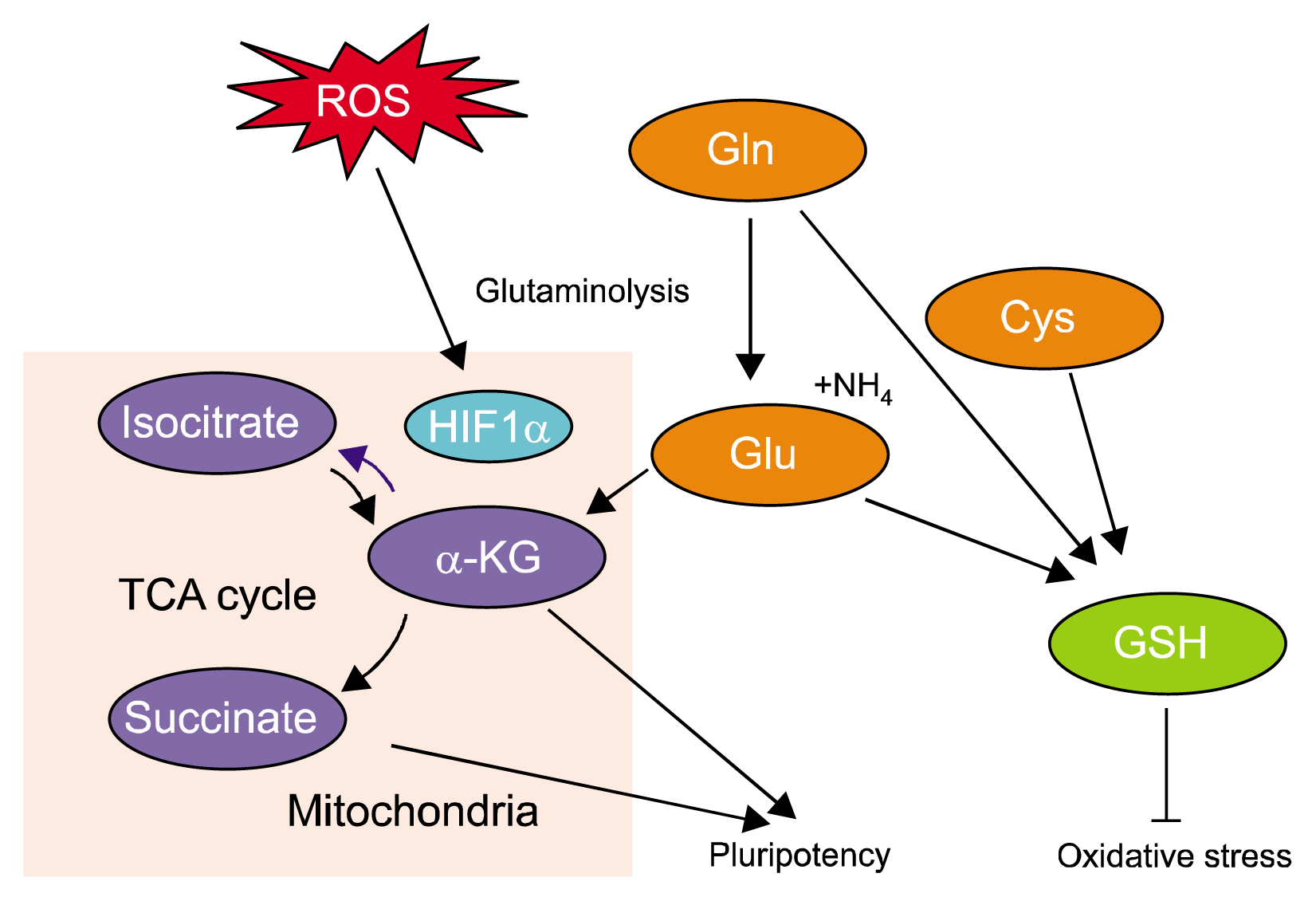
Although glucose is generally regarded as a major substrate for mammalian cells, other nutrients, including amino acids and fatty acids are also metabolized into the intermediates of the metabolic pathway and therefore may drive energy production. In addition, it is clear that glycolytic metabolism closely interacts with that of other nutrients which indicates that stem cells have distinct amino acid metabolic profiles as seen in glycolytic metabolism, and this metabolic pathway could be a critical factor in determining stem cell fate (84–87). Among the amino acids, a relationship between glutamine (Gln) and oxidative stress in the regulation of stem cell function has been reported. Gln is the most abundant amino acid in the blood and acts as a secondary energy source for the anabolic process. Gln was also identified as being essential for fuelling mitochondrial metabolism in rapidly dividing cancer cells (88–90). Furthermore, human newborns with a congenital Gln synthetase deficiency presented with developmental disorders such as brain malformation, multiple organ failure, and infant death (91, 92), which indicates that Gln has an important role in embryo development. Gln is divided into glutamate and ammonia through glutaminolysis which is initiated by deamination of Gln by glutaminase. And then, glutamate is further converted through a second deamination step into a TCA cycle intermediate, α-ketoglutarate (α-KG) by glutamate dehydrogenase (93). α-KG can be oxidized to succinate (standard TCA cycle reaction) or reductively carboxylated to isocitrate (reverse TCA cycle) through α-KG dehydrogenase (α-KGDH), respectively. This reductive cycle of glutamine metabolism has been shown to be favored in cells where HIF-1α is stabilized (94, 95). In neuronal progenitor cells, FoxO3-regulated enzymes involved in central carbon metabolism act as regulator of ROS by controlling the flow of glucose and Gln carbon in a defined metabolic pathway. FoxO3 has an indispensable role in metabolic adaptation under stress conditions by inhibiting hypoxia-induced ROS production and the subsequent HIF-1α stabilization by antagonizing c-Myc function (96, 97). In addition, Gln metabolism also regulates cellular oxidative stress through antioxidant synthesis. The availability of amino acids such as Gln, glutamate, and cysteine are involved in the biosynthesis of cellular GSH, but the NADP+/NADPH levels control the oxidative state of GSH (Fig. 3) (89, 95, 98). More recently, mouse ESCs were cultured in 2i medium (containing GSK3β and MAPK inhibitors) which maintained the naïve pluripotency and proliferation even without Gln and represented the high α-KG to succinate ratio. In addition, direct manipulation of the intracellular aKG/succinate ratio is enough to regulate pluripotency-associated gene expression through histone modification and Tet-dependent DNA demethylation (87). These previous results suggest that Gln metabolism contributes to the prevention of oxidative stress and the regulation of stem cell fate through antioxidant synthesis and alteration of the metabolic pathway in a FoxO3-dependent manner (99). As well as glutamine, high glucose-induced ROS stimulates leucine and proline uptake in mouse ESCs, which suggest that these amino acids are also implicated in the regulation of ROS-mediated stem cell function (100). Furthermore, glutamine-derived citrate can be transported to the cytoplasm to generate acetyl CoA for anabolic processes such as fatty acid synthesis (94, 101), which indicates that nutrient metabolisms are closely associated with each other; therefore, the regulation of the metabolic pathway could be important in controlling stem cell fate.
Effect of ROS on fatty acid metabolism in stem cells
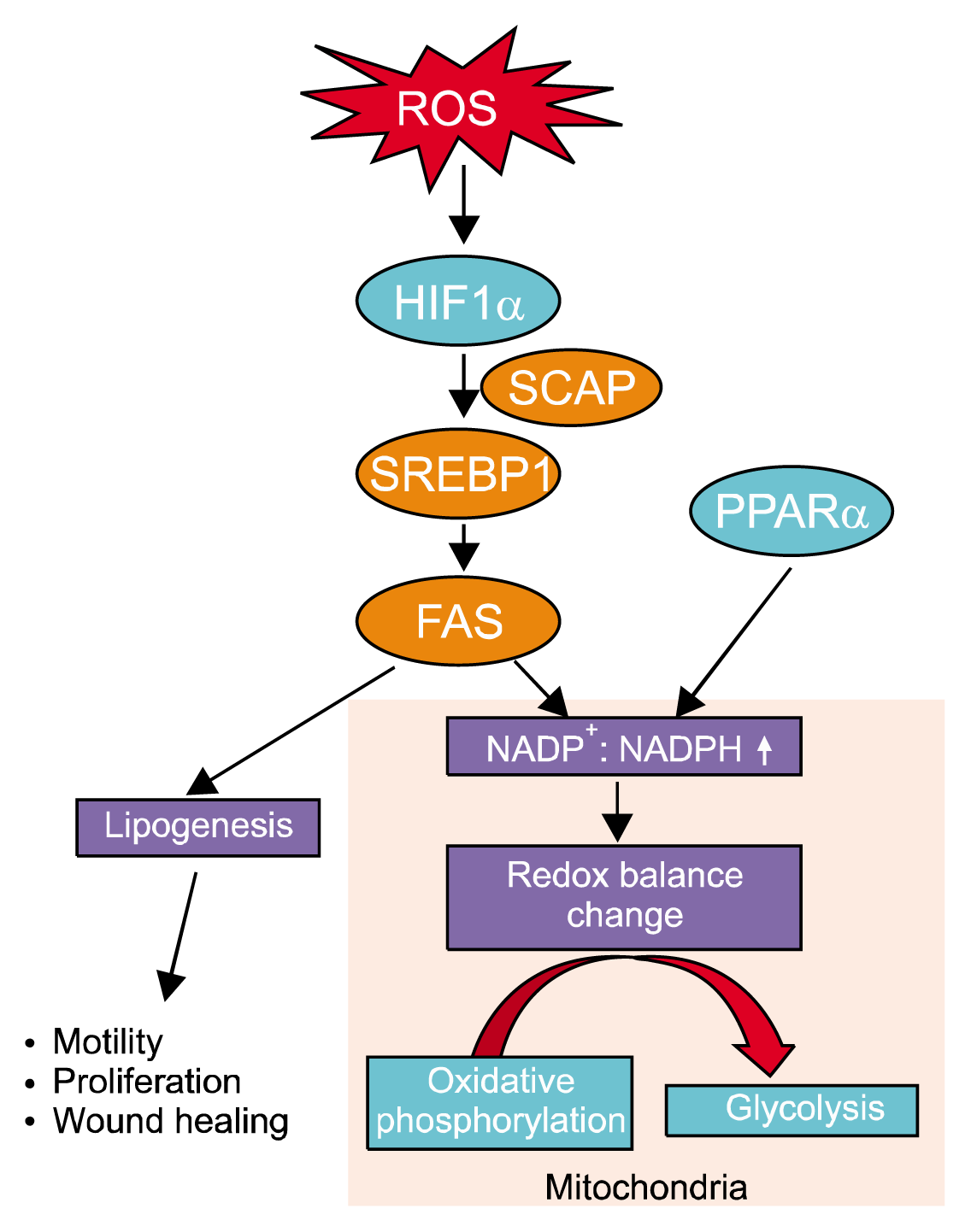
Fatty acids with long chains of lipid-carboxylic acid act as energy source and membrane components, such as phospholipids and glycolipids. They also have critical roles in the maintenance of normal cellular function and homeostasis as well as acting as signal molecules in the regulation of proliferation and differentiation (102). Fatty acid synthase (FAS), a key enzyme in de novo lipogenesis, condenses acetyl-CoA and malonyl-CoA to produce palmitic acid with NADPH as a cofactor (103). FAS pathway regulates redox homeostasis through its ability to consume reducing equivalents such as NADPH (104). A previous report provided evidence that hypoxia regulates lipid metabolism, and its metabolites are associated with determining stem cell fate. In that study, hypoxia increased HIF-1α along with SREBP cleavage activating protein (SCAP1) and sterol regulatory-element binding protein 1 (SREBP1) expression. SREBP has been known to be a key transcription factor in regulating lipogenic genes including the FAS gene (105). Hypoxia-induced FAS expression subsequently increased palmitic acid production, which stimulated human mesenchymal stem cell (hMSC) motility and increased the therapeutic effect of stem cells in a mouse wound healing model (106). In addition, hypoxia was shown to cause an increase of the NADH+/NADPH ratio due to altered metabolic flux from oxidative phosphorylation to glycolysis, which appears to have a role in adipogenic differentiation of MSCs (107). Activation of FAS stimulates lipogenesis which uses more NADPH and alters the redox balance to compensate for the shortage of oxygen. Recently, many reports have shown that oxidative stress induced FoxOs expression (108, 109) is involved in the control of adipogenic differentiation (Fig. 4) (110, 111). Consistently, during adipogenesis, the cells are likely to increase antioxidant enzyme expression, which supports the resistance to oxidative stress and subsequently helps to avoid cellular damage or apoptosis (112, 113). In addition, peroxisome proliferator-activated receptor α (PPARα), a member of the nuclear receptor protein group, could be activated by ligands including several fatty acids (114). PPARα has crucial roles in the regulation of many physiological functions such as vascular tone, inflammation and glucose homoeostasis (115). In addition, it has been reported that the activation of PPARα is needed for cardiogenesis of stem cells, which is associated with ROS-dependent metabolism (56). Similarly, a previous study has shown that PPARα negatively regulates ROS derived by NOX expression, particularly regulatory subunit p47phox, which affects the number of BM-resident EPCs and the differentiation state of monocytic progenitor cells (116). Moreover, increasing oxygen utility to generate energy from metabolites to support the energy demand according to the specialized functions of differentiated progeny implies the generation of more reduced metabolites. Interestingly, it has been reported that saturated metabolites from differentiation medium promotes differentiation, while unsaturated fatty acid impairs lineage specification through the inhibition of the eicosanoid pathway (117). Among the lipid oxidation products of ROS-dependent reactivity, there are molecules generated by the oxidation of cholesterol, polyunsaturated fatty acids and phospholipids, such as lysophospholipids, platelet activating factor-like lipids, isoprostanes, sphingolipids and ceramides (118). These ROS-dependent oxidized lipids and lysophosphatidic acids suggest the possibility that bioactive lipids such as oxycholesterol, sphingosine-1-phosphate, and lysophosphatidic acid could be associated with the regulation of stem cell functions in their hypoxic niche (119–121).
Conclusion
Currently, stem cells have attracted much attention due to their distinct features that support infinite self-renewal and differentiation into cellular derivatives of three lineages. These features suggest that stem cells have the potential to provide effective treatments for a wide range of human diseases. Recent studies have suggested that many stem cells both embryonic and adult stem cells reside in specialized niches defined by hypoxic condition. Moreover, metabolic changes accompany stem maintenance and self-renewal, which might be brought about by signals that influence stem cell fate. To deliver on the promise of stem-cell therapy, there is a need to increase our fundamental understanding of how ROS generation is regulated in stem cells and what are the exact mechanisms in which ROS determine stem cell fate through metabolic pathway alteration. Our current understanding shows that the regulation of ROS has a vital role in maintaining the stemness and differentiation of stem cells through metabolic pathway alteration.
 XML Download
XML Download