

 PDF
PDF Citation
Citation Print
Print


Introduction
Induced pluripotent stem cells (iPSCs) generated by reprogramming of fully differentiated somatic cells from patients into self-renewing pluripotent stem cells has completely transformed our ability to study disease in human cells (1, 2). Using iPSC technology, it is possible to create both genetically inherited and sporadic diseases in vitro (3). Moreover, since iPSCs can be derived directly from patients with a given disease, they carry all of the genetic components associated with the disease, which make certain cell types more susceptible to a disease state, thereby serving as the most useful genetic model of the disease (4).
Huntington’s disease (HD) is a devastating neurodegenerative genetic disease, caused by abnormal expansion of CAG repeats in the huntingtin (HTT) gene (5). Clinical features of HD include progressive motor dysfunction, cognitive decline, and psychiatric disturbance, probably caused by both neuronal dysfunction and neuronal cell death (6). Pathological hallmark of HD is the formation of aggregated huntingtin inclusion in the cytoplasm and nucleus, characterized by the expression of EM48 (7, 8). Despite numerous efforts, there are no available therapeutic options for HD at the moment (9).
We recently reported the characteristics of a HD patient-derived iPSC carrying 72 CAG repeats (HD72-iPSC) (10, 11). In this study, we investigated the in vivo roles of HD72-iPSC in the YAC128 transgenic mice, carrying the entire 170-kb genomic locus of human HTT gene plus 25 kb of the 5’ flanking sequence and about 120 kb of 3’ flanking sequence, which is commonly used as a pre-clinical model in HD (12). To do this, we transplanted HD72-iPSC-derived neural precursors into the striatum of YAC128 mice bilaterally and observed a significant behavioral improvement in the grafted mice. Interestingly, the transplanted HD72-iPSC-derived neural precursors formed GABAeric neurons efficiently, but no EM48-positive protein aggregates were detected at 12 weeks after transplantation. Taken together, these results indicate no HD pathology was developed from the grafted cells, or no transmission of HD pathology from the host to the graft occurred at 12 weeks post-transplantation, which will provide useful insights on the in vivo function and pathogenic effects of HD patient-derived iPSCs.
Materials and Methods
Culture and neuronal differentiation of HD72-iPSC
We cultured and maintained HD72-iPSC according to the method described previously (10). Neuronal differentiation was induced by co-culturing HD72-iPSC with PA6 stromal cells (obtained from RIKEN Cell Bank, Ibaraki, Japan) (13). Using the 5-stage protocol, we initially induced neural rosette-like structures, detached them mechanically and formed neurospheres in suspension culture. To make neural precursor cells, we dissociated neurospheres we dissociated neurospheres following treatment with Accutase™ (Chemicon, Temecula, CA, USA) and plated onto poly L-ornithine (PLO; 15 μg/ml, Sigma, Sigma, St.Louis, MO, USA)/fibronectin (FN; 1 μg/ml, Sigma)-coated 60 mm2 tissue culture dishes. Neural precursor cells were maintained in GMEM supplemented with 1% penicillin, 1% streptomycin, 1% nonessential amino acids, 0.1% β-mercaptoethanol, N2 supplements and 20 ng/ml bFGF (Invitrogen, Carlsbad, CA, USA).
YAC128 transgenic mice and cell transplantation
We performed animal experiments in accordance with the CHA University IACUC (Institutional Animal Care and Use Committee; IACUC090012). YAC128 mice (FVB-Tg(YAC128)53Hay/J) were originally purchased from the Jackson Laboratory (Bar Harbor, ME, USA), and 12 month-old mice were used in transplantation experiments. We stereotaxically injected a total of 15 mice with 1μl of HD72 iPSC-derived neural precursor cells (100,000 cells/μl) into the striatum of both hemispheres using the following coordinates: AP +0.5 mm, ML ±1.75 mm and DV −2.5 mm from the Bregma. In the sham group (n=15), 1μl of suspension medium (GMEM) were injected into both hemispheres in parallel. Uninjected YAC128 wild-type littermates were included as a control group (n=14). Both cell-transplanted and medium-injected animals were received cyclosporine A intraperitoneally (5 mg/kg, Sigma) 24 h before transplantation and daily up to 12 weeks.
Behavioral test
Rotarod test was performed, in which mice were placed on the rod with an accelerating rotating speed from 4 to 40 rpm over a period of 3 minutes with a 15 minutes rest between trials (14). Mice were trained on three consecutive days for three trials per day before transplantation. Three trials were performed and the mean latencies to fall were used to analyze data.
Statistical analysis
We performed statistical analyses on the rotarod scores using the Statistical Analysis System (Enterprise 4.1; SAS Korea) on a CHA University mainframe computer. Performance measures were analyzed using the PROC MIXED program, a linear mixed models procedure. A p value of <0.05 was considered significant.
Immunohistochemistry
To investigate the pathological features of YAC128 mice, we sacrificed the 12 month-old YAC128 mice and their wild-type littermates, perfused and fixed their brains with 4% paraformaldehyde. 30 μm frozen coronal sections were prepared using a cryostat (Microm, Walldorf, Germany). A conventional DAB (3,3’-diaminobenzidine) immunohistochemistry using antibodies for DARPP-32 (1:100, Cell Signaling Technology, Danvers, MA, USA) and EM48 (1:50, Chemicon) was performed. To analyze the marker expression from the transplanted cells, we sacrificed the transplanted mice at 12 week-post transplantation and performed fluorescence immunohistochemical analyses using the following primary antibodies: human-specific nuclei (hNu) (1:200, Chemicon), human-specific mitochondria (hMito) (1:200, Chemicon), human-specific Nestin (hNestin) (1:200, Chemicon), MAP2 (1:200, Chemicon), GAD6 (1:200, Developmental Studies Hybridoma Bank, Iowa City, IA, USA), Gamma-aminobutyric acid (GABA) (1:5000, Sigma), DARPP-32 (1:100, Cell Signaling), SVP38 (1:200, Sigma), EM48 (1:50, Chemicon). Secondary antibodies used were goat anti-mouse IgG-conjugated Alexa-555 (1:200, Molecular Probes), goat anti-rabbit IgG-conjugated Alexa-488 (1:200, Molecular Probes, Eugene, OR, USA) and goat anti-mouse IgM-conjugated Alexa-555 (1:200, Molecular Probes). We counter-stained the cells using DAPI, and photographed the staining patterns using a confocal laser-scanning microscope imaging system (LSM510, Carl Zeiss, Inc., Thornwood, NY, USA).
Results
Pathological features of YAC128 transgenic mice
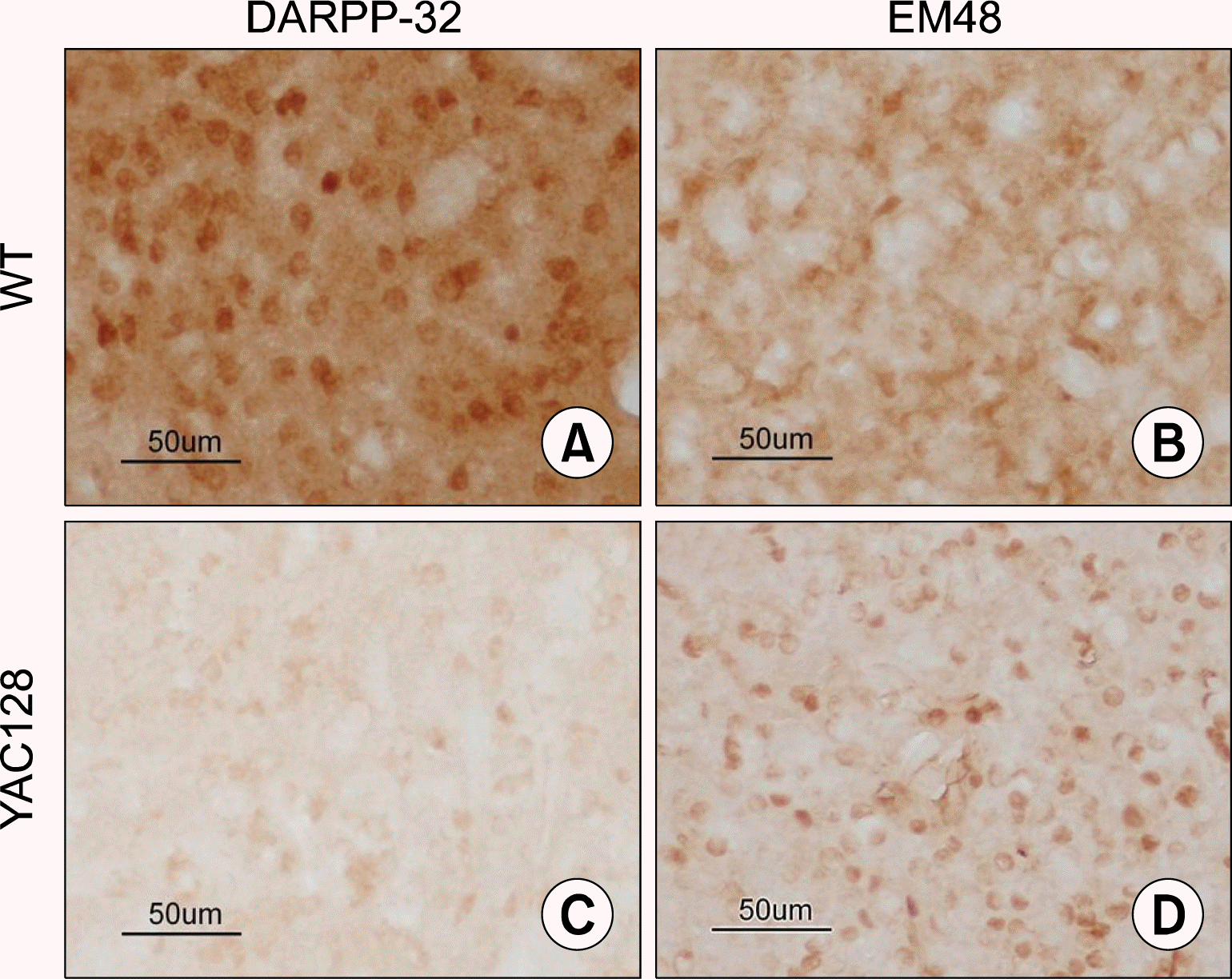
We first examined the pathological characteristics of 12 month-old YAC128 transgenic mice, by which full-spectrum of neuronal loss and behavioral deficits are known to become evident (12). As expected, we observed the expression of DARPP-32, a marker for medium spiny neurons (MSNs) is significantly reduced, whereas the expression of EM48, a marker for aggregated mutant huntingtin protein (mHtt) is significantly increased in the transgenic mice (Fig. 1B and D, respectively), compared with their wild-type littermates (Fig. 1A and C, respectively).
Behavioral improvement following transplantation of HD72-iPSC-NP cells into YAC128 mice
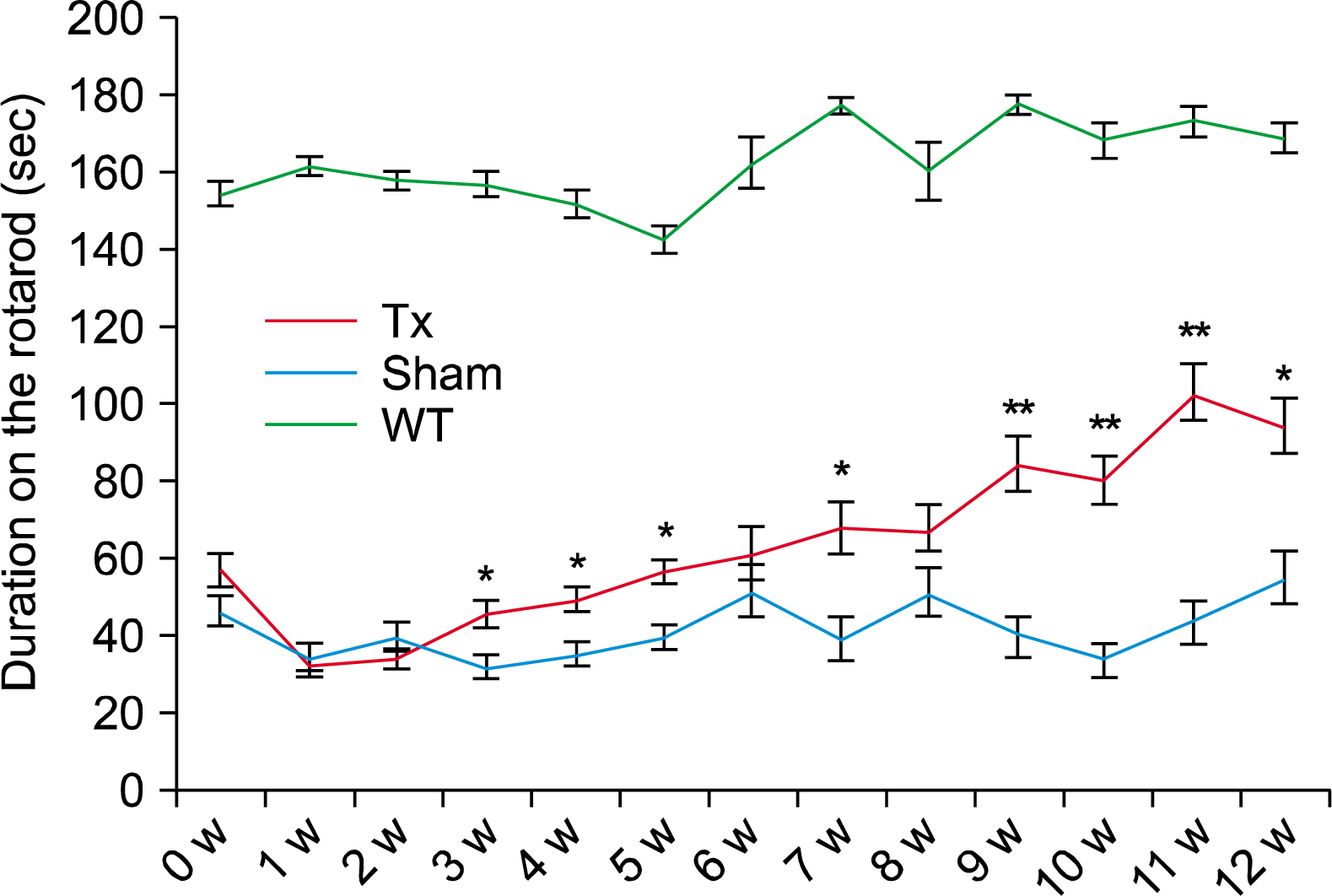
To address the in vivo roles of GABAergic neurons derived from HD72-iPSCs (10), we transplanted 100,000 HD72-iPSC-derived neural precursors, in a volume of 1μl, into the striatum of both hemispheres of each YAC128 mice (n=15). In sham control mice (n=15) we only injected suspension medium (GMEM) into both hemispheres in parallel. Uninjected YAC128 wild-type litter-mates were included as a control group (n=14). To evaluate functional effects of the grafts, we employed rotarod test, because it has been shown that rotarod performance is greatly reduced in the YAC128 mice. They exhibit rotarod deficit initially at 4 months of age but become more prominent at 6 months of age (12). Poor performance on the rotarod represents motor learning deficits, indicating some aspects of cognitive functions are also affected in YAC128 mice. To our surprise, we observed a significant improvement of rotarod activity in the transplanted group at as early as three weeks after transplantation, compared with the sham controls (Fig. 2). Therefore, these results strongly suggest that HD72-iPSC-derived neural precursors played some beneficial roles after transplantation, although they carry genetic mutations.
Neuronal differentiation and pathology development from the transplanted HD72-iPSC-NPC
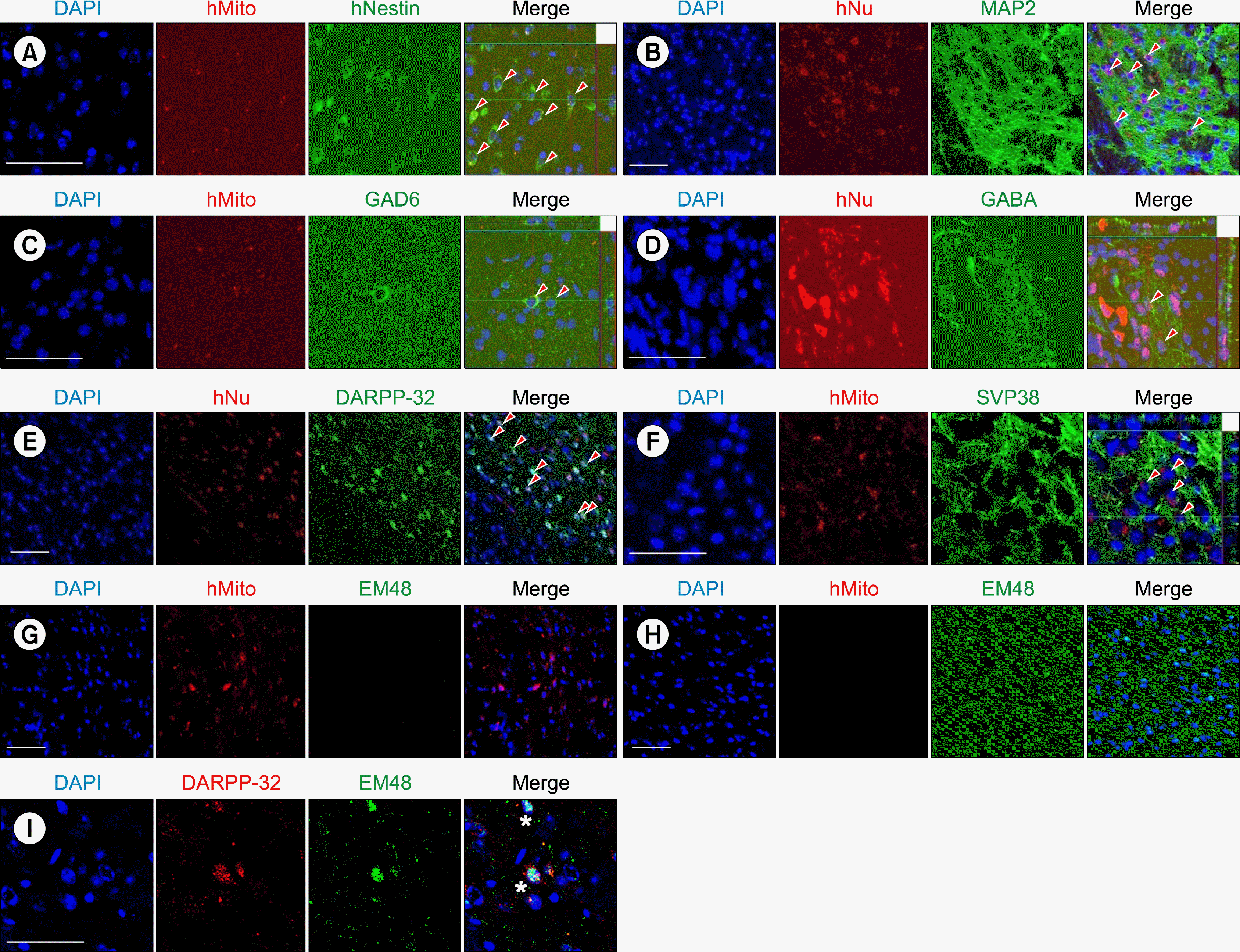
After completion of behavior test at 12 weeks, we sacrificed the mice and analyzed their brains histologically. To identify the transplanted human cells, we used antibodies against human-specific nuclear antigen (hNu) and human mitochondria (hMito). Using confocal microscopy and double immunostaining, we found that some of the transplanted HD-iPSC-derived cells were still Nestin-positive neural precursors (Fig. 3A). Importantly, a large number of the surviving human neurons were MAP2-positive (Fig. 3B), and some neurons exhibited GABAergic and MSN features, i.e., they were immunoreactive for GAD6 (Fig. 3C), GABA (Fig. 3D) and DARPP-32 (Fig. 3E). We also found that transplanted human cells expressed the synaptic vesicle protein synaptophysin (SVP38) (Fig. 3F), suggesting the possibility of synapse formation in the graft.
Intranuclear inclusions of huntingtin are a pathological hallmark of HD, which can be readily detected using an antibody against aggregated huntingtin (EM48) (Ref 20, 21). We examined whether HD72-iPSC-derived neurons develop huntingtin aggregates 12 weeks after transplantation. Interestingly, we found no EM48-positive aggregates were formed from the transplanted HD72-iPSC-derived neural precursors at this stage (Fig. 3G), while the host tissue in the YAC128 mice normally expresses EM48 (Fig. 3H). In this case, we also observed that DARPP-32-positive medium spiny neurons were already affected by EM48-positive aggregates (Fig. 3I).
Discussion
In this study, we investigated the in vivo roles of HD72-iPSC in the YAC128 transgenic mice, a commonly used preclinical mouse model of HD, carrying the entire 170-kb genomic locus of human HTT gene (12). To do this, we transplanted HD72-iPSC-derived neural precursors into the striatum of YAC128 mice bilaterally and observed a significant improvement in the rotarod performance. Interestingly, although they carry genetic mutations, the transplanted HD72-iPSC-derived neural precursors formed GABAeric and MSN-type neurons efficiently, and led to no obvious neuropathological development, judged by the absence of but EM48-positive protein aggregates at 12 weeks after transplantation. This observation was very similar to our recent report (10), in which the same type of cells were transplanted into the quinolinic acid-lesioned rodent model of HD. However, in the same report, we also showed that HD pathology can develop after ∼9 months in vivo (10), suggesting that cellular age may be a key factor in identifying aggregates in human HD-iPSCs (4). Therefore, it will be likely that the transplanted HD72-iPSC-derived neural precursors will give rise to EM48-positive aggregates at later stages. Another important aspect of results is that no EM48-positive aggregates were detected in the transplanted cells at 12 weeks post-transplantation, while the host tissue in the YAC128 mice normally expresses EM48. Moreover, DARPP-32-positive medium spiny neurons were already affected by EM48-positive aggregates. These results strongly indicate that no HD pathology was developed either from the transplanted cells, or no transmission of HD pathology from the host to the graft took place at 12 weeks post-transplantation. It will be interesting to investigate whether “host-to-graft” or “graft-to-host” transmission of HD pathology can occur at later stages. At the same time, it will be important to address whether and how normal iPSC-derived GABAergic neurons can contribute to the functional recovery and neuronal regeneration. In order these HD-iPSCs to be used for the cell therapy, it will essential to target the HTT mutation and correct it to a normal gene in the HD-iPSCS before transplantation (15). There are a wide range of HD-iPSC lines available through HD iPSC consortium (16), so it will be useful to uncover their pathogenic mechanisms in the future. Taken together, results from this study can provide useful insights on the in vivo function and pathogenic effects of human HD-iPSCs, as well as on the development of HD therapeutics in the future.
 XML Download
XML Download