

 PDF
PDF Citation
Citation Print
Print


Introduction
Driving human embryonic stem cells (hESCs) along specific differentiation pathways remains a formidable challenge for the development of hESCs-based therapy. There are several signaling pathways, such as Notch, Hedgehog (Hh), BMP, and Wnt that have been identified during early development (1–4). Especially Notch signaling has been shown critical for the maintenance of stem cells, however, it also has a role in the specification of cell fate and other aspects of regulation of differentiation (5–7).
The Notch pathway is evolutionarily conserved and the basic molecular players are receptors (Notch1 to 4), ligands (Delta, Serrate, and Jagged), and the transcription factors (8, 9). Upon ligand binding, the signaling is initiated by a proteolytic cleavage of Notch by γ-secretase and release of the Notch intracellular domain (NICD). NICD subsequently translocates to the nucleus, where it modulates expression of downstream genes (10, 11). It has been demonstrated that NICD-dependent transcriptional activity, and thus the stem cell pluripotency, may be regulated by oxygen partial tension (11). It is interesting to note that several previous studies confirmed that peri-cellular oxygen concentration has an effect on hESCs self-renewal and differentiation (12, 13). In the case of hematopoietic stem cells that reside in a relatively hypoxic niche, it has previously been shown that activation of Notch blocked differentiation and resulted in T cell acute lymphoblastic leukemia (14). However, deletion of Jagged and Notch did not result in depletion of hematopietic stem cells or disruption of hematopoiesis (15). It is thus plausible that stem cells residing in microenvironment with low availability of oxygen may require another pathway to fully activate Notch target genes that inhibit differentiation, thereby contributing to stem cell self-renewal.
Considering a possible crosstalk, Sonic hedgehog has been shown to act on the target cells to increase the transcription of several genes, including members of the Notch (16, 17). Previous studies have shown that upon binding of Hh proteins to its receptor Patched (Ptc), the Smoothened is activated, which is followed by the processing and activation of Gli transcription factors that enter the nucleus to control differentiation processes during early and late embryogenesis (18). Moreover, Hh signaling was recently suggested to play an important role in the control of ESCs growth (19). At present, it is not clear to what extent the enhanced maintenance of hESCs differential potential in hypoxia is supported by Notch and Hh signaling. In the current study, we therefore set out to examine the effect of these two pathways on the self-renewal of hESC line CLS1 that has continuously been propagated in an atmosphere with 5% oxygen concentration.
Go to : 
Materials and Methods
Cell culture
The hESC line CLS1 (20) was cultured in 35 mm tissue culture dishes (Nunc, Roskilde, Denmark) on a confluent monolayer of γ-irradiated human foreskin fibroblasts (HFF) (American Type Culture Collection no. SCRC-1041). The culture medium consisted of the Knockout Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 20% Knockout Serum Replacement, 1 mM L-glutamine, 0.1 mM β-mercaptoethanol, 1% nonessential amino acids, penicillin/ streptomycin (all Invitrogen, Carlsbad, CA), and 4 ng/ml recombinant human bFGF (BioSource Europe, Nivelles, Belgium). Cells were sub-cultured in four-week intervals and transferred in a humidified hypoxic cell culture facility with an atmosphere of 5% O2 and 5% CO2 buffered with nitrogen (Xvivo System; BioSpherix, Redfield, NY). The medium was changed every other day. HeLa cells (ATCC no. CCL-2) was used as a control and cultured in Eagle’s Minimum Essential Medium, supplemented with 10% fetal bovine serum, glutamin, and antibiotics (Invitrogen) at 37°C in 5% CO2.
To inhibit Notch and Hh signaling, γ-secretase inhibitor (γ-SI) (L1790; Sigma-Aldrich, Brøndby, Denmark) and cyclopamine (CP) (C4116; Sigma-Aldrich) were used, respectively. They were dissolved in DMSO and applied to achieve 1 μM final concentration for γ-SI and 10 μM concentration in the case of CP. The inhibitors were incubated with CLS1 cultures for a period of three weeks.
Immunocytochemistry
In order to visualize the nuclear DNA, Hoechst 33342 (Invitrogen) was added to the cultures to achieve a final concentration of 10 μg/ml and the incubation was continued at 37°C for 30 min. The cells were then washed and fixed with 4% buffered formaldehyde at 4°C for 20 min. After additional washing and blocking with 2% bovine serum albumin (BSA) at room temperature (RT) for 25 min, the cells were incubated with a 200-fold diluted SSEA1-specific mouse monoclonal antibody (sc-21702; Santa Cruz Biotechnology, Santa Cruz, CA) at RT for 1 h followed by a 200-fold diluted Alexa 488 goat anti-mouse antibody (Invitrogen) at RT for 35 min. To detect Oct4, the cells were first permeabilized with 0.2% Triton X-100 in 4% BSA at RT for 20 min, and then incubated with Oct4-specific antibody (ab19857; Abcam, Cambridge, United Kingdom) diluted 1:300 at RT for 1 h. The intra-nuclear complexes of primary antibody were reacted with Cy-5 donkey anti-goat conjugate (AP130S; Millipore, Bedford, MA) diluted 1:150 at RT for 35 min. Cells were finally washed and stored in PBS at 4°C. All washings and dilutions were done with phosphate buffered saline (PBS) and 1% BSA in PBS, respectively. The images were acquired using Axio Observer Z1 (Carl Zeiss, Göttingen, Germany) wide-field fluorescence system equipped with AxioCam MRm camera (Zeiss) and using three-channel full resolution tiled images based on 10-fold magnification. The histomorphometric analysis was carried out with the aid of AxioVision software package (Zeiss).
Bromodeoxyuridine incorporation
To monitor cell proliferation, the cultures were incubated with 10 μM bromodeoxyuridine (BrdU) (Sigma-Aldrich) at 37°C for 1 h in a CO2 incubator, washed, and fixed with 4% buffered formaldehyde at RT for 20 min. After additional washing, the proteins were denatured with 1 N HCl at RT for 30 min, and to minimize un-specific antibody binding, a blocking with 2% BSA for 30 min was carried out. The cell membranes were then permeabilized with 0.2% Triton X-100 in 4% BSA for 20 min and subsequently washed, after which an incubation with anti-BrdU mouse monoclonal antibody (A-21300, Invitrogen) diluted 1:20 in 1% BSA followed at RT for 1 h. Binding sites of primary antibody were visualized with a 200-fold diluted Alexa 488 anti-mouse conjugate (A-11001, Invitrogen) at RT for 30 min. The cells were then counter-stained with Hoechst 33342 diluted to 10 μg/ml final concentration. After final washing, the cells were examined and images taken using Axio Observer. Two-channel full resolution tiled images were acquired based on 10-fold magnification.
[3H]-Thymidine incorporation
The [3H]-thymidine incorporation experiments were carried out according to Chen et al. (21) with some modifications. Briefly, the medium was removed and replaced with fresh Knockout DMEM containing, in addition to all supplements, 825 μCi/mL thymidine [methyl-3 H]-tri-phosphate ammonium (TRK 242; Amersham Biosciences, Piscataway, NJ). The incubation proceeded at 37°C for further 4 h in a CO2 incubator, after which, the cells were washed four times with PBS, released from the feeder layer, and solubilized in 100 μL of 0.5% SDS at 37°C during 30 min. The proteins were precipitated with an equal volume of trichloroacetic acid (TCA) at RT during 10 min incubation and recovered at 1000 rpm (110g RCF) for 10 min. The pellets were dissolved in 100 μL of 1 N sodium hydroxide, transferred to 3 mL of scintillation liquid (Ultima Gold; Perkin Elmer, Waltham, MA), and the levels of β-radiation were determined using a liquid scintillation counter (RackBeta; Perkin Elmer).
Real-time RT-PCR
The total RNA was extracted using the Aurum Total RNA Mini Kit (Bio-Rad Laboratories, Hercules, CA) according to manufacturers′ recommendations and the yield was determined spectrophotometrically (ND-1000; Nano-Drop Technologies, Wilmington, DE). cDNA synthesis was performed on 200 ng of RNA using iScript cDNA synthesis kit (Bio-Rad Laboratories). For amplification reaction, an 8 μl cDNA aliquot was applied together with 13 μl of iQ SYBR Green Supermix (Bio-Rad Laboratories) reaction components, and 0.03 pmoles of 18S primers or 0.192 pmoles of gene-specific primers in a total volume of 25 μl in translucent microtiter plates (iCycler iQ PCR plates; Bio-Rad Laboratories). The samples were run on MyIQ single-color real-time PCR detection system (Bio-Rad Laboratories) using a two-step amplification cycle. The profile consisted of a single annealing/extension step of 30 sec at 60ºC and a denaturation step of 15 sec at 95ºC, for a total number of 40 cycles. All primers were designed with DNASTAR Software package (DNASTAR, Madison, WI) and were custom synthesized by DNA Technology A/S, Aarhus, Denmark (Table 1). Prior to the gene-specific analysis, the samples were evaluated for the contents of 18S rRNA that served as an internal endogenous standard. After normalization with respect to the 18S rRNA, the relative transcriptional levels were determined for each sample and gene in a semi-quantitative fashion by referring to a standard dilution series prepared from a pooled sample using MyIQ software (Bio-Rad Laboratories). To confirm the quality of each run, the occurrence of primer dimers was monitored by invoking a melting curve function of the program. All samples were analyzed in duplicates, with individual replicates being placed remotely from each other.
Table 1.
List of primers for real-time RT-PCR
![]()
Western blot analysis
The designated colonies of CLS1 line were removed, washed, and lysed in 50 mM Tris [pH 7.5], 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 0.5% NP-40, 1 mM Na3VO4, 0.5 mM PMSF, 5 mM β-mercaptoethanol, 1x protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) on ice for 30 min and the lysates were subsequently cleared at 14,000 rpm (21.910 g RCF) at 4°C for 15 min. The protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories) and samples amounting to 20 μg were separated by SDS-PAGE using a 10% gel. The transfer to a nitrocellulose membrane (Millipore) was done by a dry electroblotting procedure (Invitrogen). After the transfer, the membranes were incubated with 5% (w/v) nonfat-milk in PBS at RT for 1 h to block non-specific binding. The primary antibodies were diluted in 1% (w/v) nonfat-milk in PBS to achieve optimal concentration, rabbit polyclonal anti-NF-κB (ab7970; Abcam) 1:1000, rabbit polyclonal anti-Oct4 (ab19857; Abcam) 1:500, and mouse monoclonal anti-β-actin (A1978; Sigma-Aldrich) 1:3000, and incubated with membranes at 4°C overnight. After washing, horseradish peroxidase-conjugated either anti-rabbit (sc-2004; Santa Cruz Biotechnology) diluted 1 :2000 or anti-mouse (P0260; DakoCytomation, Glostrup, Denmark) diluted 1:1000 were incubated with membranes at RT for 1 h, and the visualization of the antibody complexes was done on the basis of enhanced chemiluminiscence (ECL Plus; GE Healthcare Life Sciences, Hillerød, Denmark) using a Kodak Image Station 4,000 MM PRO (Carestream Molecular Imaging, Woodbridge, CT). Densitometric analysis was performed with the aid of an integrated Kodak MI software package (Carestream Molecular Imaging).
Statistical analysis
All experiments were performed at least twice, and the data are presented as means+standard error of mean (SEM). Comparison of independent samples was done with a Student’s t-test and statistical significance was assigned to differences with p<0.05.
Go to :
Results
Accelerated differentiation of hESCs exposed to 5% oxygen through inhibition of Notch and Hh signaling
We have previously reported that Notch signaling is necessary for the maintenance of pluripotency of hESCs exposed to hypoxic conditions (13). Therefore we sought to investigate whether inhibition of Notch and Hh pathways has the potential to promote differentiation alone or in combination at 5% oxygen concentration. To this end, we employed γ-SI to block formation of NICD and CP to inhibit Hh signaling. The differentiated hESCs were identified through their lack of Oct4 and presence of SSEA1. The immunofluorescence staining revealed a significant induction of SSEA1 in the centers of the colonies at the expense of Oct4-positive area irrespective of whether the inhibitors were used individually or simultaneously (Fig. 1A). Using histomorphometric analysis, it was established that the extent to which the hESCs self-renewal was inhibited, in terms of the proportion of undifferentiated colony areas, was similar when γ-SI and CP were used separately, 48% and 56%, respectively (Fig. 1B). However, their simultaneous application resulted in enhanced inhibition, with undifferentiated zones corresponding only to 36%. In all cases the inhibition was statistically significant.
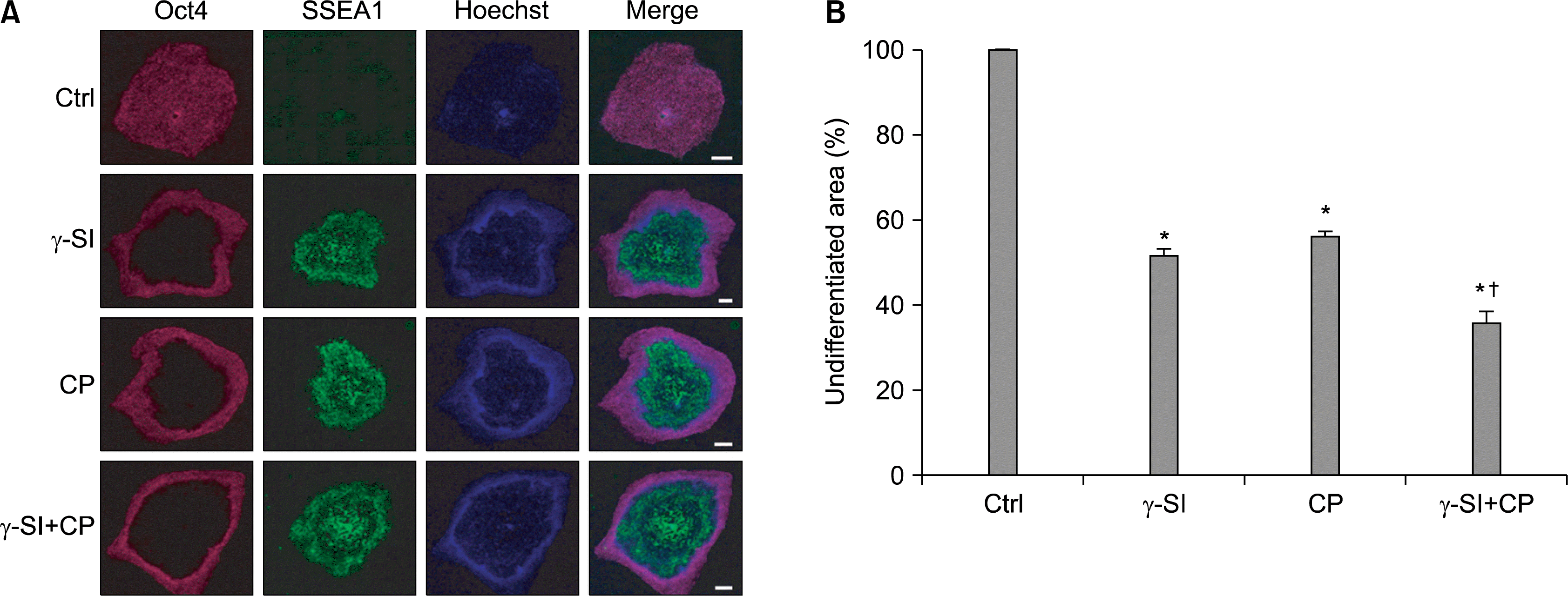 | Fig. 1.Effect of Notch and Hh signaling inhibition in hESCs exposed to 5% oxygen. (A) The hESC line CLS1 was treated with 1 μM γ-SI, 10 μM CP or combination thereof for 3 weeks. In the representative image, the undifferentiated and differentiated colony zones were distinguished by the expression of Oct4 and SSEA1, respectively. The scale bars indicate 1 mm. (B) The stained colonies were evaluated by a histomorphometric analysis. The values are presented as the mean and the error bars denote the standard error of mean. *Indicate statistical significance (p<0.05) with respect to the control, †Indicates statistical significance (p<0.05) of the combined treatment compared with single inhibitors.
|
The pattern of Oct4 distribution as identified by in-situ immunodetection was further confirmed by a real-time RT-PCR (Fig. 2A) and western blotting (Fig. 2B). These trials demonstrated that the transcriptional and translational expression resulting from inhibition closely resembled the overall reduction of undifferentiated colony areas. Confirming the loss of pluripotency in cells incubated with γ-SI and CP, another marker of pluripotency, the transcription factor Nanog, showed a practically identical decrease of mRNA expression (Fig. 2C).
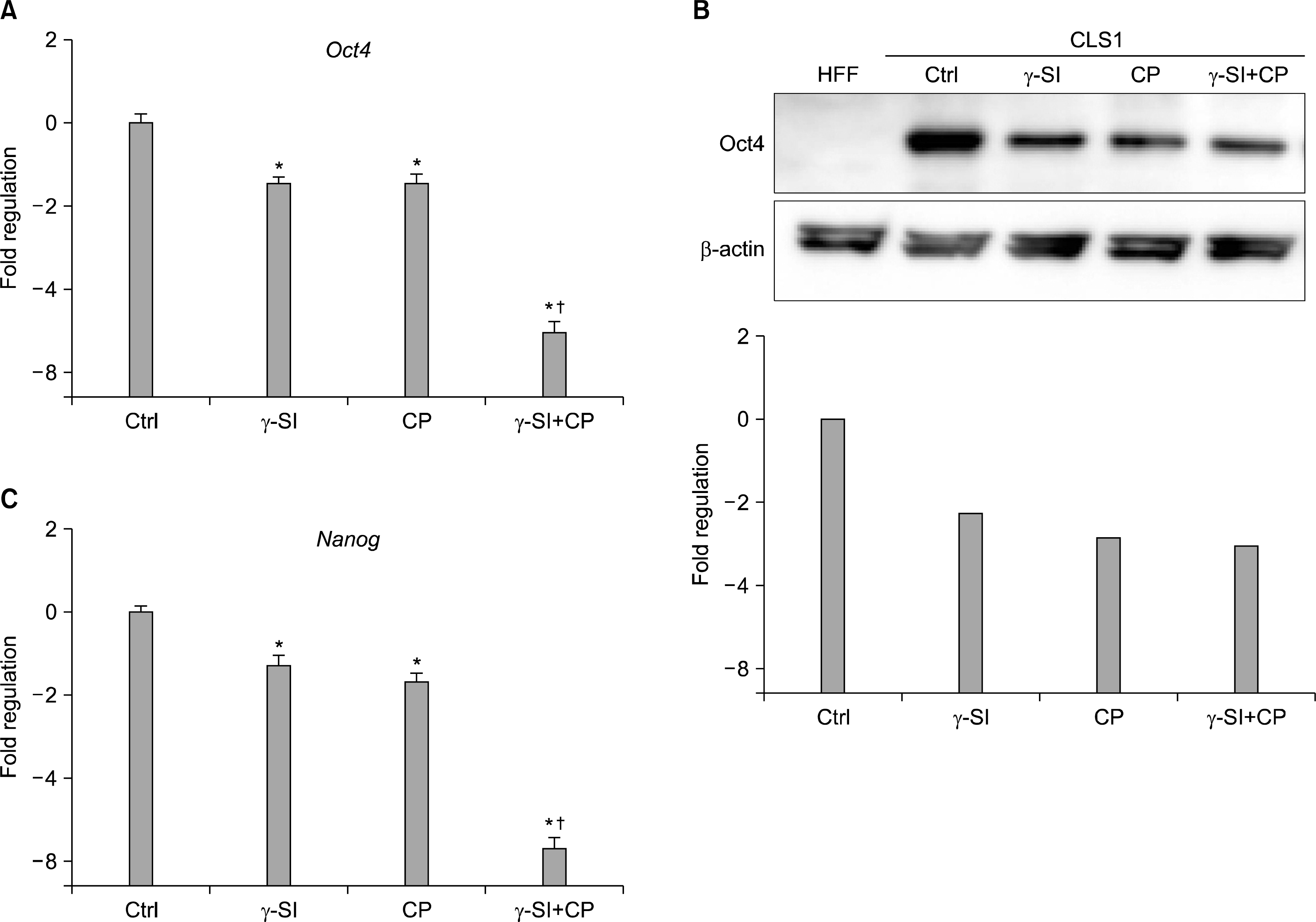 | Fig. 2.Expression of pluripotency-associated markers in hESCs exposed to 5% oxygen after inhibition of Notch and Hh signaling. The hESC line CLS1 was treated with 1 μM γ-SI, 10 μM CP or combination thereof for 3 weeks and the expression of specific genes was studied at the transcriptional and translational levels. (A) The presence of Oct4-specific messages was analyzed by a real-time RT-PCR. (B) The presence of Oct4-specific protein was analyzed by western blotting. Beta-actin was visualized for the purpose of normalization. (C) The levels of Nanog-specific mRNA were determined by a real-time RT-PCR. The values are presented as the mean and the error bars denote the standard error of mean. The normalization was done with respect to the control samples. *Indicate statistical significance (p<0.05) in comparison to the control, †Indicate statistical significance (p<0.05) of the combined treatment compared with single inhibitors.
|
Effect of Notch and Shh inhibition on downstream genes
As our initial experiments implicated Notch and Hh signaling in mediating differentiation of hESCs exposed to 5% oxygen, we next sought to investigate whether expression of the Hes1 and Hey1, and Gli1 and Ptc1 immediate downstream genes (22, 23), respectively, was altered in our system. The analysis of transcriptional activation by a real-time RT-PCR of colonies grown for three weeks in hypoxic conditions has interestingly shown a significant decrease of both Hes1 and Hey1 mRNA not only with γ-SI but also with CP and combined γ-SI+CP (Fig. 3A). Similarly, when analyzing the levels of Gli1 and Ptc1 transcripts, not only the Hh specific inhibitor CP demonstrated a significant effect but also the application of γ-SI, and γ-SI in combination with CP resulted in a significant suppression (Fig. 3B). It is noteworthy that the simultaneous action of γ-SI and CP seemed to have a significant additive effect.
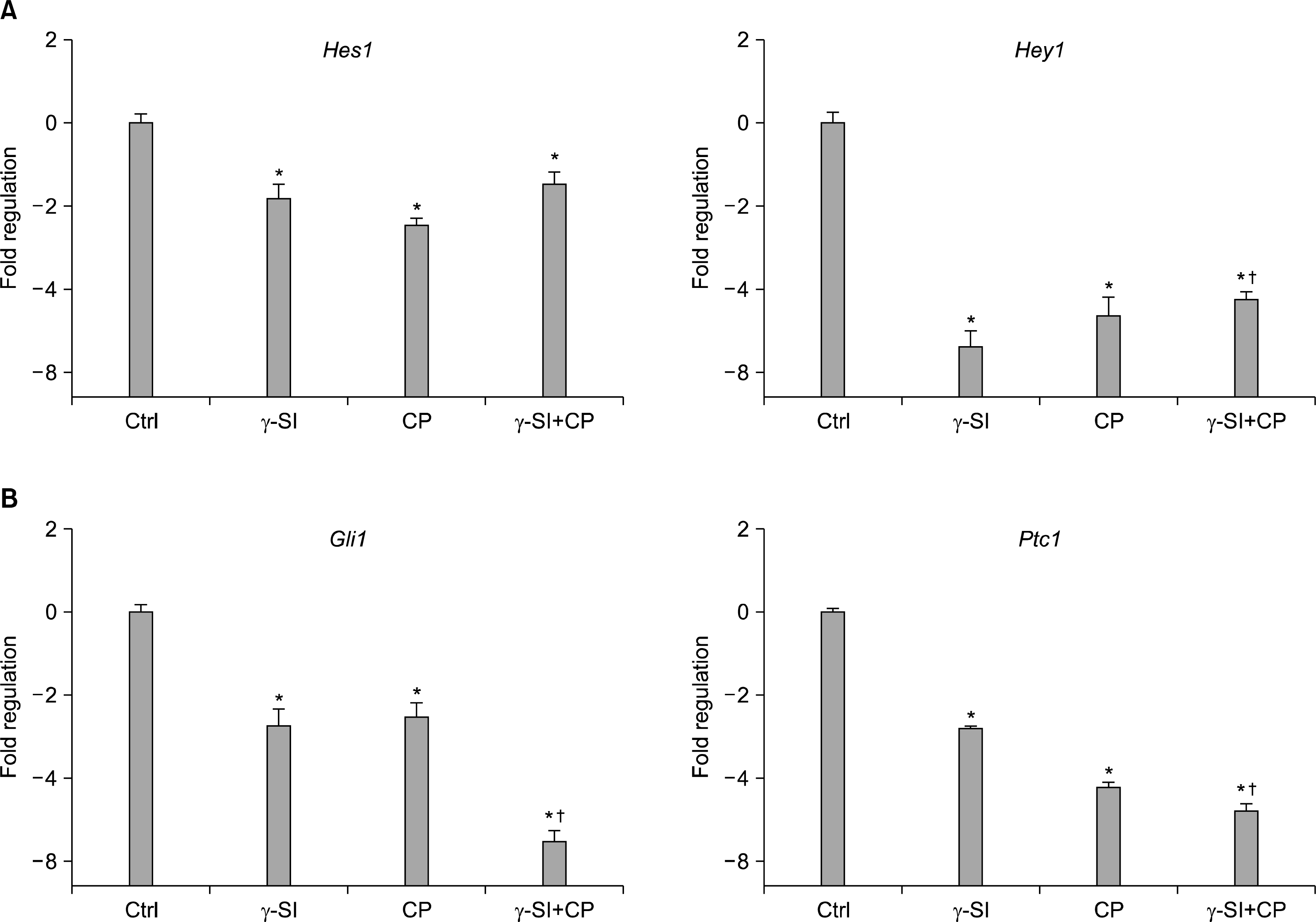 | Fig. 3.Transcriptional activation of downstream genes after Notch and Hh inhibition in hESCs grown in 5% oxygen. The hESC line CLS1 was treated with 1 μM γ-SI, 10 μM CP or their combination, and after 3 weeks the levels of gene-specific messages were determined by a real-time RT-PCR. (A) The expression of Notch effector genes Hes1 and Hey1. (B) The expression of Hh effector genes Gli1 and Ptc1. The values are presented as the mean and the error bars denote the standard error of mean. The normalization was done with respect to the control samples. *Indicate statistical significance (p<0.05) in comparison to the control. †Indicate statistical significance (p<0.05) of the combined treatment compared with single inhibitors.
|
hESC proliferative responses to inhibition of Notch and Hh signaling
Since the modulation of differentiation potential is associated with altered proliferative capacity, we investigated the effect of γ-SI and CP inhibitors on this parameter using BrdU and [3H]-thymidine incorporation assays, and NF-κB western blot analysis. BrdU was instrumental in identifying regions in the colonies, where the cell division was affected. As shown in Fig. 4A, the treatment with either γ-SI, CP or both resulted in a characteristic inhibition of proliferation in the central areas of the colonies. From comparison with Fig. 1A, it is obvious that these areas are coincident with the inhibition of self-renewal. Again, it is apparent that the simultaneous use of inhibitors produced a more pronounced effect than each of them individually. Quantitative assessment of the inhibition of proliferation was based on [3H]-thymidine incorporation (Fig. 4B). These results correspond well with the observed changes in colony morphology after BrdU visualization in panel A.
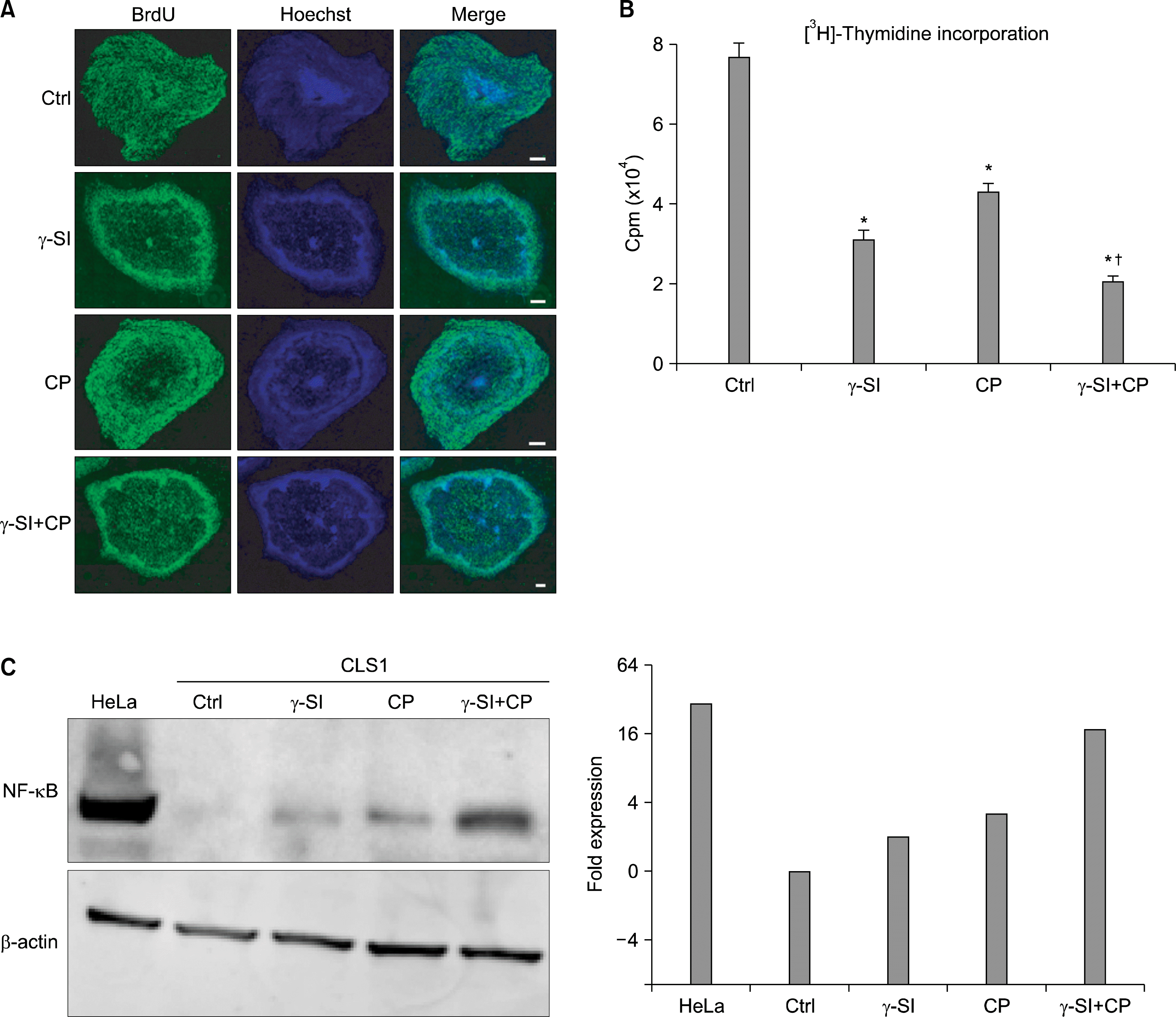 | Fig. 4.Proliferative potential of hESCs maintained in hypoxia (5% O2) in the presence of inhibitors of Notch and Hh signaling. The hESC line CLS1 was treated with 1 μM γ-SI, 10 μM CP or their combination for 3 weeks. (A) Representative image demonstrating incorporation of BrdU. The scale bars indicate 1 mm. (B) [3H]-thymidine incorporation. The values are presented as the mean and the error bars denote the standard error of mean. (C) Western blot analysis of NF-κB. Beta-actin was visualized for the purpose of normalization and the plotted data were normalized to the control. *Indicate statistical significance (p<0.05) with respect to the control, †Indicates statistical significance (p<0.05) of the combined treatment compared with single inhibitors.
|
As for the expression of NF-κB, the protein levels increased as a result of inhibition with each of the individual inhibitors (2.6-fold on average) (Fig. 4C). Interestingly, however, the most significant increase occurred after the combined application (17.9-fold).
Go to :
Discussion
hESCs are defined as cells with self-renewal capacity (24). It is becoming clearer that both self-renewal and differentiation in stem cells involve numerous interdependent pathways with considerable crosstalk and that various factors, including oxygen availability, have a significant role in these interactions (1, 4, 11). We have recently shown that hESCs can be maintained in un-differentiated state at 5% oxygen for up to 18 months without spontaneous differentiation (13). In the present report, we searched for the signaling components in un-differentiated hESCs exposed to low oxygen and asked whether these components interact to maintain the hESCs self-renewal. We found that both γ-SI, an inhibitor of Notch signaling pathway, and cyclopamine, an inhibitor of Hh signaling pathway, induced hESC differentiation that resulted in increased levels of SSEA1 marker. This potent inhibitory effect also resulted in reduced numbers of hESCs in S phase as shown by [3H]- thymidine and BrdU incorporation. Highly interestingly, we found that the employed inhibitors did not have a selective effect on the specifically targeted pathway, rather they inhibited, with a comparable efficiency, the unrelated pathway as well. This cross-inhibition provides evidence for a tight cooperation between Notch and Hh signaling and highlights the significance of both pathways in oxygen-mediated control of hESCs identity.
Previous studies have reported that Notch signaling is critical for the maintenance of stem cells (5–7) and that Hh signaling plays a role in the control of embryonic stem cell growth (19). Recent evidence indicates that the control of the fate of hESCs is accomplished through cooperative effect of multiple factors (25, 26). No studies have been performed so far to investigate the combined effect of Notch and Shh pathways in the maintenance of hESCs during exposure to low oxygen. In the current investigation, the simultaneous treatment of hypoxic hESCs with both inhibitors resulted in a significantly greater inhibition of undifferentiated cells than when γ-SI or CP was used alone. It is important to note that although an additive effect was observed, the achieved inhibition was still not complete. This suggests that multiple pathways in parallel are required to keep enhanced pluripotency of hESCs exposed to low oxygen and that Notch and Hh have complementary role in this process, in addition to other factors. Further studies are necessary to provide a thorough mechanistic explanation for this interaction but it is important to note that the inhibition experiments appear to indicate that both pathways converge on the same, at least herein identified, set of downstream genes. Of special interest would be to reveal basis for a crosstalk with hypoxia-mediated signaling.
In order to further our knowledge on the maintenance of hESCs exposed to low oxygen, we searched for the expression of nuclear factor-kappaB. NF-κB is an important transcription factor that controls the expression of a wide variety of genes involved not only in innate and adaptive immunity, but also in cell proliferation and apoptosis (27, 28). In the present study, we found enhanced expression of NF-κB in hESCs exposed to low oxygen resulting from inhibition of Notch and Hh signaling. We have not analyzed the mechanism(s) leading to enhanced expression of NF-κB, what leaves the need for more research in this direction. However, constitutive NF-κB expression has been noted in many tumors, including colorectal cancer, and found to play key role in angiogenesis and anti-apoptosis, thus promoting tumor growth (29). Over the past decade, a number of experiments have been performed that support the idea that cancers can grow from a discrete subpopulation of malignant cells with stem cell properties (30, 31). Moreover, in hematopoietic tumor cells, such as BCL10 and MALT1, NF-κB is expressed and has been implicated in the pathogenesis of mucosa-associated lymphoid tissue lymphomas (32).
The present results show that Notch and Hh pathways play a cooperative role in the maintenance of hESCs pluripotency when exposed to low oxygen concentration. Although additional investigations will be required to obtain complete understanding of the involved processes at the molecular level, our findings provide additional insight in the regulatory events underpinning enhanced hESC self-renewal in hypoxia.
Go to :
 XML Download
XML Download