

 PDF
PDF Citation
Citation Print
Print


Regenerative medicine is seeking for an innovative therapeutic strategy that assures to ameliorate health and quality of life by restoring or regenerating cells, tissues or organs. Cellular sources of regenerative medicine are an essential matter. Cells derived from adult, umbilical cord, fetus and embryonic origin have been shown therapeutic potentials (1, 2).
Even though, pluripotent embryonic stem (ES) cell which gives rise to cells from all three germ lineages seems to be the most idyllic candidate for regenerative medicine, these pluripotent stem cells could be removed from growing embryos, resulting in ethical concerns. In adults, stem cells or progenitor cells can be taken from the tissue that contains stem cells, such as bone marrow, heart, brain, skin, muscle, adipose tissue, eyes, kidneys, lungs, liver, gastrointestinal tract, pancreas, breast, ovaries, prostate and testis (3–10). The tissue-specific adult stem cells can derive new more differentiated and specialized cells and thus repopulate the tissues in which they live beneath homeostatic environments as well as regenerate injured tissues after severe injuries. However, adult tissue-derived stem cells possess innate limitation, in terms of stem cell potency and therapeutic potential.
Since the ideal source of stem cells for potential therapeutic purposes remains controversial, stem cell researchers look for pluripotent stem cell that could be isolated from the adult tissues or generated from already differentiated cells (11, 12). Exact pluripotent stem cell should possess both potential for multi lineage differentiation in vitro and more importantly, also is capable to harmonize in vivo blastocyst development. Generation of pluripotent stem cells from already differentiated cells or somatic cells is called dedifferentiation and/or reprogramming. Reprogramming could be defined that it takes normal adult body cells such as skin cells and sends each cell's nucleus back to a pluripotent state. In other words, the reprogrammed cells would then be capable of producing any tissue type in the body essentially equivalent in versatility to ES cells. They could then be used to grow tissues for future use in regenerative medicine. For example, these reprogrammed cells could be used for treating numerous genetic and degenerative disorders. Among them, age-related functional defects, hematopoietic and immune system disorders, heart failures, chronic liver injuries, diabetes, Parkinson’s and Alzheimer’s diseases, arthritis, and muscular, skin, lung, eye, and digestive disorders as well as aggressive and recurrent cancers could be successfully treated by stem cell-based therapies (3, 7, 9, 13–15).
The reprogrammed pluripotent cells could be a perfect genetic match: these cells would not be rejected by the donor's immune system. Most importantly, there would be no embryo created, destroyed, damaged or used in any way at any point in the process. In addition, ethicists might be more favorable to this type of regenerative medicine as opposed to embryonic stem cells. Here, we review and summarize recent breakthroughs and limitations to generate pluripotent stem cells from somatic cells and their potential applications in regenerative medicine.
Stem cells
Stem cells are characterized by the ability to renew themselves through mitotic cell division and differentiating into a diverse range of specialized cell types. Stem cell development begins with the totipotent zygote which is able to differentiate to any type of tissues in the body including the placenta. The blastocyst forms after seven to eight cell divisions of the fertilized egg. Blastocyst outer wall is modified to hold fast to the uterine wall and the inner cell mass (ICM) contains pluripotent cells that are able to differentiate all types of tissues and organs within the developing fetus. These are defined as ES cells. In 1981, ES cells were first isolated from mice and human ES cell lines were established in 1998 (16, 17). ES cells were differentiated to variety of multipotent stem cells and lineage-specific cells. For example, hematopoietic stem cells (HSCs) are multipotent cells. HSCs further differentiate to form all types of blood cells but it can not differentiate to form other cell types (18).
Adult stem cells are present in almost all the organ tissues (1, 3–10). If any damages are occur in adult tissues, it can be regenerated themselves, because most of the differentiated tissues have a significant degree of homeostatic renewal, including the epidermis, liver, small intestine, and bone marrow. Every adult tissues have a small compartment of prehistoric stem cells that are able to self renew and can give rise to mature, differentiated adult cells of multiple lineages. It is also promising that occupant adult stem cells can stimulate reprogramming in adjacent committed cells to obtain a more prehistoric regenerative response. Once activated, occupant stem cells have been suspected to be active through the production of progenitor amplifying cells (19), fusion with differentiated adult cells (20) and induction of somatic cell nuclear reprogramming (21).
Stem cell based-therapies in regenerative medicine
Stem cell based therapeutic applications are a potential and quickly emerging branch of regenerative medicine in which cell-based therapy could be applied to treat and cure various hostile and fatal diseases (13, 14, 22–26). Many latest researches carried out with in vitro or ex vivo differentiated ES cells, fetal and umbilical cord blood (UCB)-derived stem cells and their functional progeny as well as adult stem/progenitor cells have provided accruing substantiation sustaining their potential therapeutic application for numerous genetic and degenerative disorders (3, 4, 14, 22, 26–33). Allogenic transplantation of stem cells or their additional differentiated progeny into patients may markedly represent a potential therapeutic approach, unaccompanied or in combination with the predictable treatments, for conquering the progressive failure of functions of adult stem cells with aging and degenerative diseases (14, 34). Besides, the genetic manipulations in ES cells or adult stem cells such as HSCs, endothelial progenitor cells (EPCs), mesenchymal stem cells (MSCs), and neural stem cells (NSCs) also present manifold potential to diminish the risk of rejection related with their in clinical applications. The genetically-altered stem cells could even be applied for reversing inherited genetic defects that are responsible for dissimilar pathological disorders (14, 35–38). Gene therapies by using genetically altered stem cells as vehicles for the delivery of therapeutic agents at precise injured organ tissues also characterize promising approaches for treating various pathological disorders and cancers (7, 14, 23, 36–39). It has been demonstrated that genetically adapted migrating NSCs, which are capable to travel through the central nervous system and reach the extracranial neoplastic sites, may be transplanted in the animal models in vivo and specifically attracted to tumor sites due to the release of chemotactic signals such as vascular endothelial growth factor (VEGF) and stromal derived growth factor-1 (SDF-1) (36, 37, 39). Recent studies have shown the potential and sustaining benefits of using stem cells or their further differentiated progeny for cell replacement therapies for treating various pathological disorders. The importance is on particular properties of tissue occupant adult stem cells and their slots found in bone marrow (BM), vascular walls, heart and brain as well as their potential therapeutic applications. However, challenging issues remain. It is not easy to acquire sufficient donor cells, and there are issues with immunological compatibility and the specific control of cell fate in defined ex vivo conditions. To overcome this problem, dedifferentiation or reprogramming of adult somatic cells would provide a perfect genetic match.
Approaches of somatic cell reprogramming
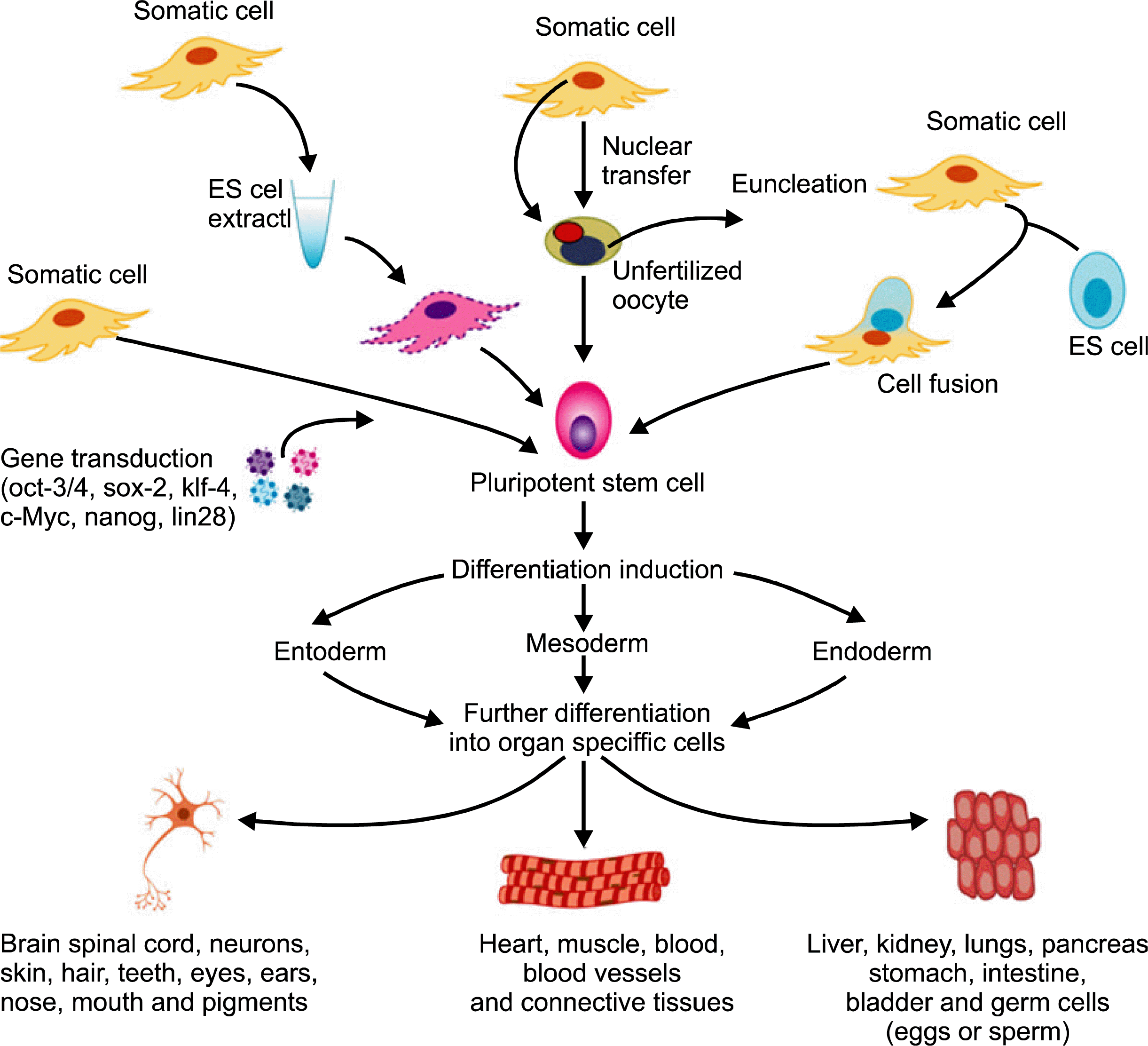
Reprogramming is a technique that involves dedifferentiation of adult somatic cells to produce donor or patient-specific pluripotent stem cells without the use of embryos. Cells generated by reprogramming would be genetically identical to the somatic cells and would not be rejected by the donor. Researchers are trying to find simpler methods with high efficiency of reprogrammed cells. This review is focusing on the representative methods for reprogramming including somatic cell nuclear transfer into oocyte, fusion with ES cell, reprogramming with pluripotency-related genes and treatment with ES cell extract (Fig. 1).
Somatic cell nuclear transfer into oocyte
In 1996, the first mammal (Dolly) was cloned by adult somatic cell nuclear transfer (SCNT), and subsequent reports on the derivation of ES cells from cloned embryos have flashed reborn enthusiasm for reprogramming to a pluripotent state. SCNT involves the removal of an oocyte nucleus in vitro, followed by its replacement with a somatic cell nucleus obtained from a donor. Cell division was stimulated by chemicals or electricity up to the blastocyst stage, at that time the inner cell mass is isolated and cultured, resulting in ES cells that are genetically identical to the donor. It has been shown that SCNT-derived ES cells, which originated from fibroblasts, lymphocytes, and olfactory neurons, are pluripotent and generate live pups after tetraploid blastocyst complementation, showing the same developmental potential as fertilized blastocysts (40–43).
The resulting ES cells are perfectly matched to the donor’s immune system, and no immunosuppressant would be required to prevent rejection. Even though ES cells derived from SCNT have the nuclear genome of the donor cells, mitochondrial DNA inherited by the oocyte could lead to immunogenicity after transplantation. Lanza et al. (44) evaluated the histocompatibilty of nuclear transfer-generated tissue; the nucleus of a bovine skin fibroblast was microinjected into an enucleated oocyte. Even if the blastocyst was implanted, the purpose was to produce cardiac, renal and skeletal muscle cells, which were then harvested, expanded in vitro and seeded onto bio-degradable scaffolds. These scaffolds were then implanted into the donor from whom the cells were cloned to determine if cells were histocompatible. The evaluation revealed that T cell response was not observed in cloned renal cells, suggesting that rejection would not essentially occur in the presence of oocyte-derived mtDNA.
However, even though promising results, SCNT has certain limitations that entail further improvement before its clinical application. First, ethical concerns about the prospective of the resulting embryos to grow into cloned embryos if implanted into a uterus should be resolved. Various animal studies have shown that blastocysts derived from SCNT can give rise to a live born infant that is a clone of the donor when implanted into a uterus. But, this reproductive cloning method is banned in most countries for human applications. In contrast, therapeutic cloning is used to derive only ES cell lines whose genetic material is matching to that of their source. In this case blastocysts are allowed to develop until a 100 cell-stage where ES cells can be acquired and thus, the blastocysts are never implanted into a uterus. Moreover, this method has not been exposed to work in humans. But, non-human primate ES cell lines have been generate by SCNT of nuclei from adult skin fibroblasts (45, 46). Second, when the SCNT-derived ES cells are using clinical application, the quality of SCNT-derived ES cells should be assessed carefully. Regrettably, one of the lines derived by SCNT discovered a translocation consisting of an isochromosome comprised of two copies of the long arm of the Y chromosome. It is not known whether chromosomal abnormalities in SCNT-derived ES cells originate from aneuploid embryos or occurred during ES cell isolation and culture. Third, the efficiency of SNCT is very low and the supply of human oocytes is insufficient, since the therapeutic potential of SCNT technique is getting delay. Taken together, SCNT has demanding restrictions to translate into clinical application.
Somatic cell fusion with embryonic stem cell
In 1976, Miller and Ruddle (47) demonstrated that thymocytes acquired pluripotency when with fused with embryonic carcinoma cells. After that, Tada et al. (48) also observed the same results by electrofusion with mouse ES cells. Transplantation of these cells into nude mice results in the formation of teratomas consisting of various tissues from all three germ layers, substantiating the pluripotency of these cells. A further progress in the cell fusion technique was reported by Cowan et al. (49) to reprogram normal diploid human fibroblasts into human ES cells. ES cells fused with the fibroblasts resulted in hybrids that sustained as predictable a tetraploid chromosome complement and displayed morphology, growth rate and surface molecule expression patterns characteristic of human ES cells. Assessment of genome-wide transcriptional changes, allele-specific gene expression and DNA methylation illustrated that the fibroblast genome was reprogrammed to near an embryonic state. Further, differentiation of ES-fibroblasts hybrids in vivo generated cell types from each germ layer. These findings showed that ES cells have the capacity to reprogram adult somatic cell chromosomes after cell fusion. ES cells may therefore provide a useful substitution of oocytes for biochemical and genetic studies aimed at understanding how to reprogram differentiated cells to an embryonic state and thereby increase their growth potential. Ultimately, this technique might lead to an alternative route for creating genetically modified-ES cell lines for use in the study and treatment of various diseases.
However, a significant technical hurdle leftover before these hybrid ES-like cells could be used for clinical applications: particularly, the abolition of the ES cell chromosomes either before or after cell fusion. Enucleation of ES cells before fusion may not be practicable in circumventing this problem as it has been revealed to eliminate the ability of the remaining ES cytoplast to re-activate expression of pluripotency markers in hybrids with somatic cells (50). The selective deletion of chromosomes is possible but may be impracticable for the complete set of chromosomes (51). Alternatively, ES cells-derived chromosomes carrying the major histocompatibilty complex loci could be removed selectively to avoid or at least reduce rejection reactions during treatment. This possibility should be experimentally investigated.
Somatic cell reprogramming using pluripotency-related genes
In 2006, a epoch-making discovery in the reprogramming of somatic cells into pluripotent stem cells has opened a new-era of regenerative medicine (52). Takahashi and Yamanaka were demonstrated that mouse embryonic fibroblasts (MEFs) and adult mouse fibroblasts can be reprogrammed into pluripotent stem cells (induced pluripotent stem (iPS) cells) (52). They analyzed 24 genes that were considered to be essential for ES cells. From them, they recognized only 4 genes, namely Oct3/4, Sox2, Klf4 and c-Myc which were essential to give ES-like properties on fibroblasts. MEFs and adult fibroblasts were co-transduced with retroviral vectors, each carrying Oct3/4, Sox2, Klf4 and c-Myc. Reprogrammed cells were identified via drug resistance. In this case, a downstream gene of Oct3/4 and Fbx15 was replaced with a drug resistance gene via homologous recombination. The consequential iPS cells possessed the immortal growth characteristics of self-renewing ES cells, expressed genes specific for ES cells and generated embryoid bodies (EBs) in vitro and teratomas in vivo. When the iPS cells were injected into mouse blastocysts they contributed to a variety of different cell types and representing their growth potential. iPS cells chosen by Fbx15 were seemingly pluripotent, but they were not indistinguishable to ES cells. Dissimilar to ES cells, chimeras of iPS cells did not effect in germline transmission. Gene expression profiles of the iPS cells demonstrated that they possessed a diverse gene expression marker compared to ES cells. The epigenetic state of the iPS cells was someplace between their somatic origins and fully reprogrammed ES cells. These results were signifying the incomplete reprogramming.
Wernig et al. (53) reported that these results were considerably improved. They infected retroviral vectors with fibroblast and preferred for the activation of endogenous Oct4 or Nanog genes. This study demonstrated that DNA methylation, gene expression profiles, and chromatic state of the reprogrammed cells were very similar to those of ES cells. Teratomas induced by these cells have differentiated cell types representing all three germ layers. Most prominently, the reprogrammed cells from the experiment were able to form viable chimeras and give to the germline-transmittable ES cells, suggesting that these iPS cells were completely reprogrammed. This might be attributed to the fact that Wernig et al. observed that the number of reprogrammed colonies increased when drug selection was initiated later. This result proposes that reprogramming is a slow and gradual process and may elucidate why the process of using Fbx15 activation on day 3 post-transfection may result in incomplete reprogramming.
Moreover, Takahashi et al. (54) and Yu et al. (55) demonstrated about the reprogramming of human cells using transduction of the 4 pluripotent genes. Yamanaka’s research group initiated by optimizing the transduction competences of human dermal fibroblasts and decided that the introduction of a mouse receptor for retroviruses into human dermal fibroblast cells using a lentivirus enhanced the transduction competence from 20% to 60%. They then demonstrated that retrovirus-mediated transfection of Oct3/4, Sox2, Klf4, and c-Myc produces human iPS cells, which are comparable to human ES cells in terms of morphology, proliferation, gene expression, surface markers, and teratoma formation (54). In dissimilarity, Thomson’s research group demonstrated that retroviral transduction of Oc4, Sox2, Nanog, and Lin28 could also produce pluripotent stem cells without introducing any oncogenes (55). These two studies were depicted that human iPS were analogous to human ES cells. However, a disquiet is that these iPS cells contain three to 6 retroviral integrations which may increase the risk of tumorigenicity. Okita et al. (56) reported that the tumor formation in chimeric mice generated from Nanog-iPS cells and observed 20% of the progeny developed tumors due to the retroviral expression of c-Myc. Another approach would be to use a brief expression method, since both Meissner et al. and Okita et al. demonstrated strong silencing of the viral controlled transcripts in iPS cells (56, 57). Their results depicts that exogenous defined factors are only required for the induction, not for the maintenance of pluripotency. One more apprehension is the use of transgenic donor cells for reprogrammed cells in the mouse studies. iPS cells were isolated by picking for the activation of a drug resistant gene inserted into endogenous Fbx15, Oct3/4, or Nanog. The application of genetically modified donors obstructs its clinical applicability for humans.
To evaluate whether iPS cells can be generated from genetically unaltered donor cells, MEFs and adult skin fibroblasts cells were retrovirally transduced with Oct3/4, Sox2, Klf4 and c-Myc and ES-like colonies were isolated by morphology, without the use of drug selection for Oct4 or Nanog (57). iPS cells from unaltered donor cells formed teratomas and generated live chimeras. This result suggests that genetically altered donor cells are not necessary to generate iPS cells. Even if this is an exciting phenomenon, it is unclear why reprogramming adult fibroblasts and mesenchymal stromal cells have analogous efficiencies (52). It would appear that cells that are previously multi-potent could be reprogrammed with greater competence, since the more undifferentiated donor nucleus the improved SCNT performs (58).
Recently Kim et al. (59) did a meaningful achievement in somatic cell reprogramming. They reported that adult mouse neural stem cells (NSCs) express higher endogenous levels of Sox2 and c-Myc compared to ES cells and then exogenous Oct4 together with either Klf4 or c-Myc is sufficient to generate iPS cells from NSCs. These two factor iPS cells are analogous to ES cells at the molecular level, contribute to development and form chimaeras. Very recently, the same group achieved another progress (60). They demonstrated that exogenous expression of the germline specific transcription factor Oct4 is sufficient to induce pluripotent stem cells from adult mouse NS cells. This Oct4 induced pluripotent stem cells are similar to ES cells in vitro and in vivo. These cells can be efficiently differentiated into neural cells, cardiomyocytes and germ cells in vitro, but they are also able to form teratoma and germline transmission in vivo. This study suggests that Oct4 is required and sufficient to directly reprogram NS cells to pluripotency (60). According to these results, for the induction of pluripotency, the number of reprogramming factors can be reduced when using somatic cells that endogenously express appropriate levels of complementing factors.
At present, researchers are trying to figure out how to prevent iPS cell-related tumor formation; it is a major concern in clinical applications (56). Nakagawa et al. (61) generated iPS cells without c-Myc from mouse and human fibroblasts, because c-Myc retrovirus was suspected to increase tumorigenicity in germline transmitted-progeny mice. This study showed significantly less non-iPS background cells and the iPS cells generated were consistently of high efficiency. Mice generated from c-Myc-free iPS cells did not show tumors during the experimental period. Also, this procedure also enabled efficient isolation of iPS cells without drug selection. In addition, this study generated human iPS cells from adult dermal fibroblasts without c-Myc.
Another disquiet in retrovirus or lentivirus-mediated iPS cells is that viral integration into the host genome may increase the risk of tumorigenicity. Hence, Stadtfeld et al. (62) generated mouse iPS cells by using non-integrating adenoviruses transiently expressing Oct4, Sox2, Klf4, and c-Myc. These adenovirus-mediated iPS cells illustrated DNA demethylation characteristics of ES cells, expressed endogenous pluripotency-related genes, formed teratomas and contribute to multiple tissues including the germ cells in chimeric mice. This result bestows strong evidence that mutagenesis with genomic integration is not required for in vitro reprogramming. The technique of non-genomic integrating reprogramming may give a better method for induction and studying patient-specific stem cells and for comparing the characteristics between ES cells and iPS cells. Besides, Okita et al. (63) also generated mouse iPS cells without viral vectors. They demonstrated that repeated transfection of a single plasmid containing the cDNAs of Oct3/4, Sox2, and Klf4, jointly with a c-Myc expression plasmid, into MEFs resulted in iPS cells without evidence of plasmid integration, which produced teratomas when transplanted into mice and contributed to adult chimeras. The generation of virus-free iPS cells, although from embryonic fibroblasts, concentrated on critical safety concern for potential use of iPS cells in regenerative medicine.
Somatic cell dedifferentiation/reprogramming using cell extracts
Reprogramming using various types of cell extracts can generate new gene-expression profiles in somatic cells. Studies using various cellular proteins or extracts have shown a modest effect on reprogramming into specific lineages (64, 65). Hakelien et al. (64) reported that 293T cells, when lymphocyte extract transferred, expressed lymphocyte markers. After that, the same group (66) demonstrated that dedifferentiation/reprogramming of 293T cells and NIH3T3 fibroblasts after incubation in extracts obtained from pluripotent cells such as embryonal carcinoma cells and ES cells. The short treatment of cells with extracts triggered the formation of colonies with a phenotypic organization of ES cells. Upregulation of a number of pluripotency genes and downregulation of somatic genes such as lamin A, were subsequently detected up to 4 weeks following treatment. Besides, these cells were able to differentiate to mesoderm and ectoderm lineages (66). The phenotypic alterations in these cells were the result of epigenetic modifications of the chromatin mediated by chromatin remodeling factors, histone acetylation and expression of specific genes and protein synthesis.
Recently Bru et al. (67) have investigated the possibility into human cells. Pluripotency-related genes expressed when human somatic 293T cells were permeabilized and incubated in extracts of mouse ES cells. Expression of all 4 genes (Oct3/4, Sox2, c-Myc and Klf4) was induced over 1 to 8 hours. Gene expression was associated with loss of repressive histone H3 modifications and increased recruitment of RNA polymerase II at the promoters. Lamin A/C, which is typically found only in differentiated cells, was also removed from the nuclei. When 293T cells were returned to culture after exposure to ES cell extract, the expression of pluripotency-related genes continued to rise over the following 48 hours of culture, suggesting that long term reprogramming of gene expression had been induced. This provides a methodology for studying the de-differentiation of somatic cells that can potentially lead to an efficient way of reprogramming somatic cells to a pluripotent state without genetically altering them. Moreover, Rajasingh et al. (68–72) also reported that, on exposure to mouse ES cells extracts by reversible permeabilization, NIH3T3 cells underwent dedifferentiation and subsequent re-differentiation by stimulus into multiple lineage cell types. However, genome wide expression profiling revealed significant differences between NIH3T3 and ES extract-treated NIH3T3 cells. Epigenetically, ES cell extracts induced CpG demethylation of Oct4 promoter, hyper-acetylation of histones 3 and 4, and decreased lysine 9 (K-9) dimethylation of histone 3 in NIN3T3 cells. This study provides evidence for the generation of functional multipotent stem-like cells from terminally somatic cells, which would represent a major step in the use of regenerative medicine.
Currently, many things still remain elusive regarding the molecular mechanisms underlying dedifferentiation by extracts or proteins. Further studies, such as which specific components of extract/protein(s) would be crucial to induce dedifferentiation and which somatic cell types would be the best candidate for dedifferentiation, are demanded.
Future directions
Due to ethical concerns, ES cells could not be used in regenerative medicine which is mainly concentrating on incurable diseases, although recently some groups have tried to apply ES cell-derived differentiated cells into clinical trials. The researchers turned towards isolation and therapeutic application of adult stem cells, umbilical cord blood-derived stem cells for regenerative medicine. But, unfortunately, the therapeutic efficiency is low as compared with ES cells. Another limitation is the difficulty in obtaining healthy autologous cells or donor cell. Also, there are issues with immunological compatibility and the precise control of cell fate in defined conditions ex vivo. To overcome current limitations, researchers have tried to generate pluripotent stem cells from somatic cells using various techniques. The dediffentiated/reprogrammed cells could be a perfect genetic match. However, current somatic cell dedifferentiation/reprogramming techniques are insufficient to generate ES-like cells: SCNT, cell fusion and cell extract treatment have some scientific, practical and ethical limitations. iPS cells generated by pluripotency-related gene using retroviruses or lentiviruses would cause tumor. To prevent the risk of tumorigenicity, researchers generated iPS cells without viral integration and reduction of oncogene. In this situation, we should find out the exact molecular mechanisms of somatic cell dedifferentiation/reprogramming. This review suggests that dedifferentiation strategies using non-viral methods such as chemical, small molecules or proteins would be the best options. Virus-free, genetic modification-free ES-like cells would be ideal for potential use in regenerative medicine.
Conclusion
This review gives an outline about stem cells and approaches of somatic cell reprogramming which are critical for regenerative medicine. We scrutinized the merits and demerits of currently available methods such as SCNT, cell fusion, viral or non-viral vectors containing pluripotent genes introduction into somatic cells to generate ES cell like cells, and cell extract treatment (Table 1). Finally, we propose that non-viral mediated dedifferentiation/reprogramming would be ideal for regenerative medicine.
 XML Download
XML Download