

 PDF
PDF Citation
Citation Print
Print


Introduction: The role of stem cells to maintain the “state of immortality” in an “mortal multicellular being”
While it might be argued that a single cell organism, such as a bacterium, is an immortal cell, during the course of biological evolution, new phenotypes emerged that provided survival advantages when cells organized into a cohesive society of cells to form a multi-celled organism. While temperature, availability of nutrients, pH, atmospheric factors, and radiation influenced the growth regulation of these single cell organisms, stability for the species was maintained by the genetic information that protected by a relatively error- free DNA repair and DNA replication system. The single cell organism species survived the inevitable changes of environment, in which it found itself, simply by being able to proliferate via a symmetrical cell division to achieve a large population, in which only a few mutations occurred, bearing on its ability to survive a new environment.
In the multi-cell organism, a “Faustian bargain”, of sorts, was made for new adaptive features that allowed this collection of cohesive cells to survive to maintain the species. Of course, while most of the factors that controlled cell growth of the single cell organism are relevant for the individual cells of the multi- cell organism, internal or endogenous growth control was needed to regulate the many cells within the multi-cell organism. Moreover, some cells gave up ordinary “self replication” ability in order to provide highly specialized adaptive function for the survival advantage of the whole organism. Thus, the process of “differentiation” appeared, together with the mechanism that allowed “asymmetric cell division” to supplement the process of symmetrical cell division. This unique feature of a cell’s ability to proliferate either by symmetrical cell division (to increase the cell numbers of like-type cells) or by asymmetric cell division (to maintain homeostatic levels of daughter-like mother cell but to allow the formation of a differentiation of a specialized cell) is a “hallmark” of a multi-cell metazoan. In addition, as a consequence of this differentiation process was the induction of “mortality” of both the specialized differentiated cell and ultimately, the whole organism. A unique form of cell death, programmed cell death or apoptosis also appeared that aided in allowing the multi-cell organism to acquire new adaptive features. The transition of the single cell, fertilized egg to a larvae, pupae and butterfly, for example, required specialized cells at each phase of development (food acquiring genes/phenotypes) to give way to newer specialized cells that ultimately provided the end-product of development of wing muscles for an adult butterfly to mate, and pass on the genes to maintain the species.
Clearly, while almost all the cells of the multi-cell organism contain the total genomic information of the species, only a portion of those genes are expressed in each specialized cell. Therefore, the process to regulate, correctly, specific battery of genes from the total genome must have emerged during this evolutionary transition from the single cell organ to the multi-cell organism.
In order that the individual, mortal, multi-cellular organism help to maintain the survival of the species to which it belonged, the formation of both germinal stem and somatic or adult stem cells was required. The relative genomic stability had to be maintained in the germ stem cells, with occasional mutations occurring, to give the species a reservoir of new genetic options to adapt to the inevitable environmental changes. Seen in this manner, the multi-cellular organism’s germinal stem cells were immortal, while the mortal individual was only the transient carrier for these immortal stem cells.
The somatic stem cell also emerged during this evolutionary transition from the single cell “immortal” organism to the mortal individual multi-cellular organism that carried immortal germ cells. In order to pass through its various development stages (embryo, fetus, neonate, adolescent, mature and geriatric stages), processes to provide the whole organism enough cells for growth, differentiation, as well as for wound-healing and death due to apoptosis, had to emerge. The development of a unique type of stem cell, which had the ability to self-renew (the two daughter cells having retained the ability be a stem cell as the mother cell), as well as to divide asymmetrically for the production of one daughter to maintain “stemness”, including maintaining immortality, and the other to be a ‘transit-amplifying” cell, committed to have a finite life span that could differentiate, senescence or apoptosis.
In contemporary terms, there seems to be a number of stem cell types. The “toti-potent” stem cell is the fertilized egg, meaning it can give rise to all cell types (approximately 200 in the case of the human being) and the reproducing individual with both the germinal and somatic stem cells. The germinal stem cell can give rise, ultimately, to either sperm or eggs. It resides in its special niche (1). As the embryo starts to form from the fertilized egg, (blastomere, gastrula, etc (2)), the toti-potent stem cell starts to restrict the daughter stem cells’ ability to give rise to a whole individual, but still maintains the ability to give rise to all the other somatic cell types. These are “pluri-potent” stem cells. As the embryo transits to the fetal stage, the micro-environmental changes, which, in all likelihood, provides different signals to regulate different genes to adapt to this new situation. Illustrating this beautiful cybernetic relationship between selectively regulating specific genes out of the total genome by a cascading self-inducing sequence as development proceeds is the description of this process by C. Markert (3):
“Cells interact and communicate during embryonic development and through inductive stimuli mutually direct the divergent courses of their differentiation. Very little cell differentiation is truly autonomous in vertebrate organisms. The myriad cell phenotypes present in mammals, for example, must reflect a corresponding complexity in the timing, nature, and amount of inductive interactions. Whatever the nature of inductive stimuli may be, they emerge as a consequence of specific sequential interactions of cells during embryonic development.
The first embryonic cells, blastomeres, of mice and other mammals are all totipotent. During cleavage and early morphogenesis these cells come to occupy different positions in the three-dimensional embryo. Some cells are on the outside, some inside. The different environments of these cells cause the cells to express different patterns of metabolism in accordance with their own developing programs of gene function. These patterns of metabolism create new chemical environments for nearby cells and these changed environments induce yet new programs of gene function in responding cells. Thus a progressive series of reciprocal interactions is established between the cellular environment and the genome of each cell. These interactions drive the cell along a specific path of differentiation until a stable equilibrium is reached in the adult. Thereafter little change occurs in the specialized cells and they become remarkably refractory to changes in the environment. They seem stably locked into the terminal patterns of gene function characteristic of adult cells. The genome seems no longer responsible to the signals that were effective earlier in development.
Of course, changes can occur in adult cells that lead to renewed cell proliferation and altered differentiation as seen in neoplasms, both benign and malignant, but such changes are very rare indeed when one considers the number of cells potentially available for neoplastic transformation. Possibly, mutations in regulatory DNA of dividing adult cells can occasionally lead to new and highly effective programs gene function that we recognize as neoplastic or malignant. However, most genetic changes in adult cells can probably lead to cell death since random changes in patterns of gene activity are not likely to be beneficial.”
As developmental processes and the internal micro-environment must become more unique, the new adult or somatic stem cell, itself, is restricted further in its ability to be a pluri-potent-like stem cell. This new adult stem cell is referred to as a “multi-potent” stem cell. These cells, in turn, can give rise to another restricted ability to give rise to different specialized cells. Again, further development limits derivatives of these multi-potent stem cells to become bi-polar stem cells, such as the “oval” cells of the liver (4). Lastly, the final restriction of these bi-polar stem cells is a “uni-polar” stem cell that only gives rise to a single differentiated progeny. The offspring of these stem cells that lose the ability to proliferate asymmetrically, but not symmetrically, are consider to be life-span limited (the “Hayflick phenomenon” (5)). These cells eventually senesce or die by terminal differentiation or apoptosis.
Over-arching all of this is the critical idea that one of the most important evolutionary developments in the emergence of the multi-cell organism is the appearance of a gene or several genes that responds to micro-environmental triggered signals that direct the cell to divide either symmetrically or asymmetrically. This might be one of the most important genes to study (6), in view of the observation that, within the stem cell theory of cancer’, dysfunction/dis-regulation of asymmetric cell division seems to be involved early in the carcinogenetic process (7). In addition, the delicate function of maintaining the “immortality” of the genome for the perpetuation of a multi-cell species has to be done within a “mortal” individual consisting of 200 terminally-differentiated cell types and some immortal germ and adult stem cells.
The state of understanding the restriction or differentiation of the toti-and pluri-potent stem cells follows a “time’s arrow” course (i.e., only one way) or whether all their progenitor-differentiated daughters can be “re-programmed” to totally de-differentiate back to the embryonic stem cell is still in a state of flux (8). While there does seem to be solid evidence of “transdifferentiation” of some cells [white fat cells to brown fat cells (9)], the evidence that this process is yet complex in the laboratory and possibly limited in vivo because of developmental and aging factors.
The concept of a stem cell being immortal is also complicated by current conceptual and experimental contradictory reports. One view has any stem cell (both embryonic and somatic/adult stems) is naturally “immortal” until it is induced to terminally differentiate or to become “mortal”. Experimentally, the observation that these stem cells seem to demonstrate genomic instability after significant cell divisions (10). This might be due to inevitable errors in replication of DNA, as the stem cells proliferate. On the other hand, this might be the consequence of inadequate in vitro culturing conditions, such as growing the cells in an oxygen rich environment (11), on substrates and micro-environmental factors different from the natural niche microenvironment found in vitro (12).
Role of stem cells in carcinogenesis: clues to stem cell biology
The origin of the concept of stem cells must have had a very long history, even though its articulation in modern terms might not be recognized. From the idea of the early Greek myth of Prometheus; of the “Perfectibility of Man” (13), to the early studies on the generation of whole plants from root tips, or regeneration of parts of the hydra, to regeneration of a limb of an amphibian, to the realization of continuous replacement/regeneration of tissues, such as the skin, lining of the intestine or blood, and to the hypothesis of the origin of cancer, the idea of a special cell in the multi-cell organism had to emerge. The fields of embryology and developmental biology had to incorporate, implicitly or explicitly, the concept of the stem cells.
In another field, i.e., the cancer researchers accepted the generalization that cancer cells were immortal and that there existed many different genotypes and phenotypes of the cells within any tumor. The image of cancer as a “disease of differentiation” (14), “cancer as a stem cell disease” (15–17), “oncogeny as partially blocked ontogeny” (18) were usually seen as supporting the idea that in the adult organism, there existed an adult stem cell. The fact that, while the individual cells within the tumor could be either or both genetic and phenotypically different, they were shown to have had their origin in a single cell. The mono-clonal origin of cancers, again, was easily incorporated into the stem cell theory (19, 20).
The link between development and cancer was also noted with various human genetic syndromes, in which the susceptibility to a teratogenic defect was linked to a high risk to some risk to specific or multiple cancers (21). In point mutation- cancer prone syndrome, such as Fanconi’s anemia (22) or xeroderma pigmentosum (23), one can find development defects. In the chromosomal mutation- associated Downs syndrome, the many developmental anomalies, are associated with only pre-mature aging and leukemia (24).
Even more evidence linking developmental defects and cancer comes from agents that can affect both teratogenesis and carcinogenesis. Of course, in the case of hereditary induction of both birth defects and cancer predisposition in the same individual by either point gene or chromosomsal mutations, it is even more interesting that many agents, that are not mutagenic, can contribute to both teratogenic process and to the carcinogenesis process. The examples of thalidomide as a well-know human teraogen (25) is now being utilized as an anti-cancer agent by blocking angiogenesis (26). Another non-mutagenic agent, retinoids, are also human teratogens (27). At the same time there is evidence these compounds can be either chemopreventive/chemotherapeutic agents (28) or they can be tumor promoting agents (29).
The question raised is: What might be a ‘shared’ underlying link between teratogenesis and carcinogenesis, as well as a link to stem cells, specific genes and to environmental/dietary agents that can influence both teraogenesis and carcinogenesis. The finding that an evolutionary- conserved set of genes, the “connexin” genes (30, 31), has been linked to stem cell biology (32), differentiation/development (33), growth control (34, 35), apoptosis (36), onco-genes and tumor suppress genes (37), and to teratogenic, tumor promoting chemicals, as well as being a shared mechanism in many chronic diseases (38–40).
Thus, abnormal gap junctional intercellular communication, based on expression and function of the connexin genes, might be that shared underlying mechanism linking teratogenesis and carcinogenesis.
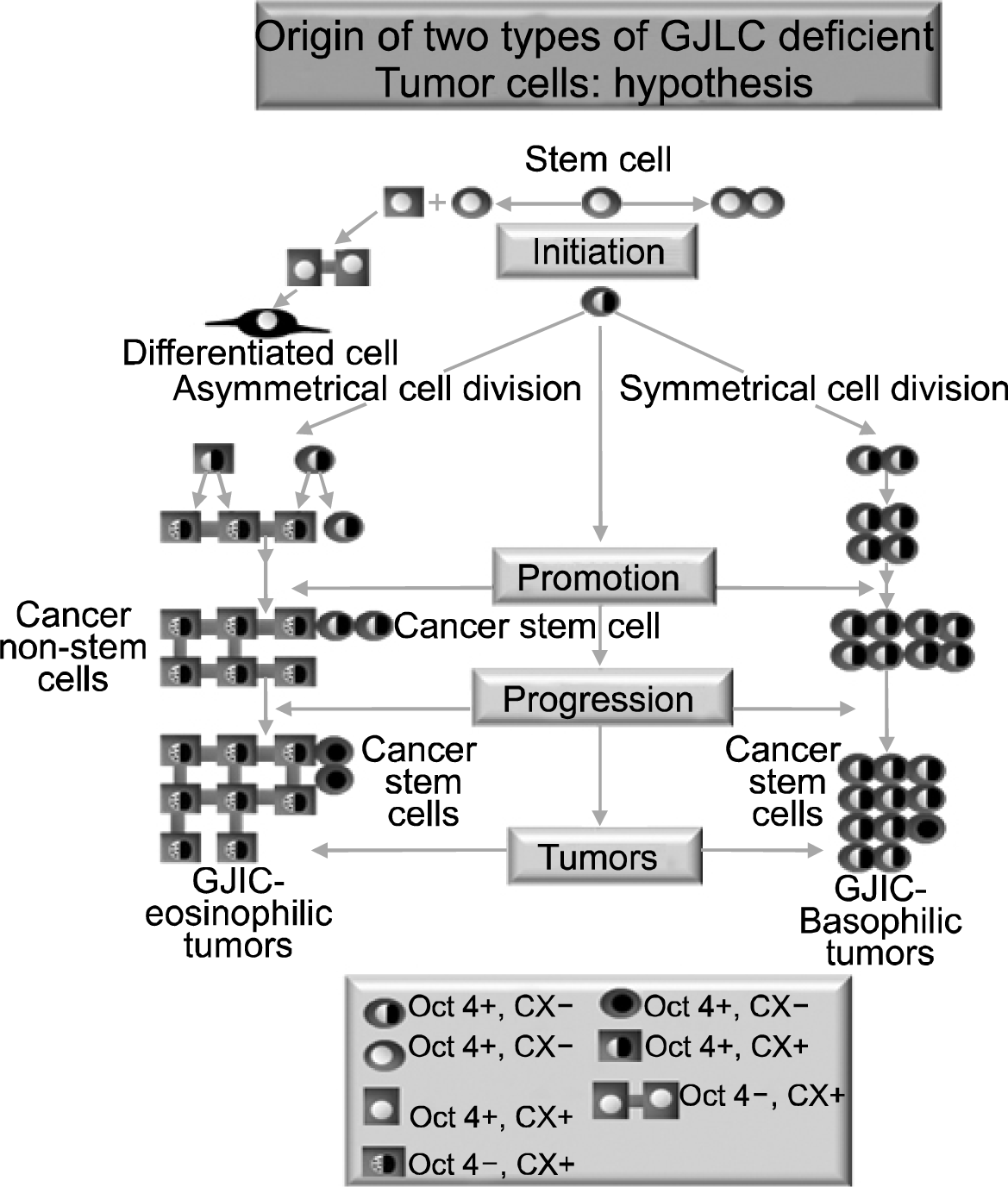
To understand the aforementioned generalizations, the major concept of the multi-stage, multi-mechanism process of carcinogenesis could help unite the stem cell concept with the process of cell-cell communication. When a single cell in an adult organism is exposed to an agent that can cause that cell to become “immortal”, it is referred to as being “initiated” (Fig. 1).
This process, operationally, is irreversible. Several animal models have supported this concept (41, 42), in addition to human epidemiology results that clearly suggest that carcinogenesis is not a 1-hit process (43). In the usually accepted paradigm, this single normal, “mortal” cell is immortalized, presumably by a mutational event in either a critical proto-oncogene or tumor suppressor gene (44). However, a stable “epigenetic” alteration has not been rigorously ruled out as being an “initiator”. In fact, given that patterns and types of human childhood cancers seem to be different from adult cancers, and given that teratomas and childhood cancers seem to be more “primitive” than the adult tumors, epigenetic mechanisms might be playing a greater role in the earlier appearances of these tumors and possibly in the more successful treatments of these tumors compared to adult tumors.
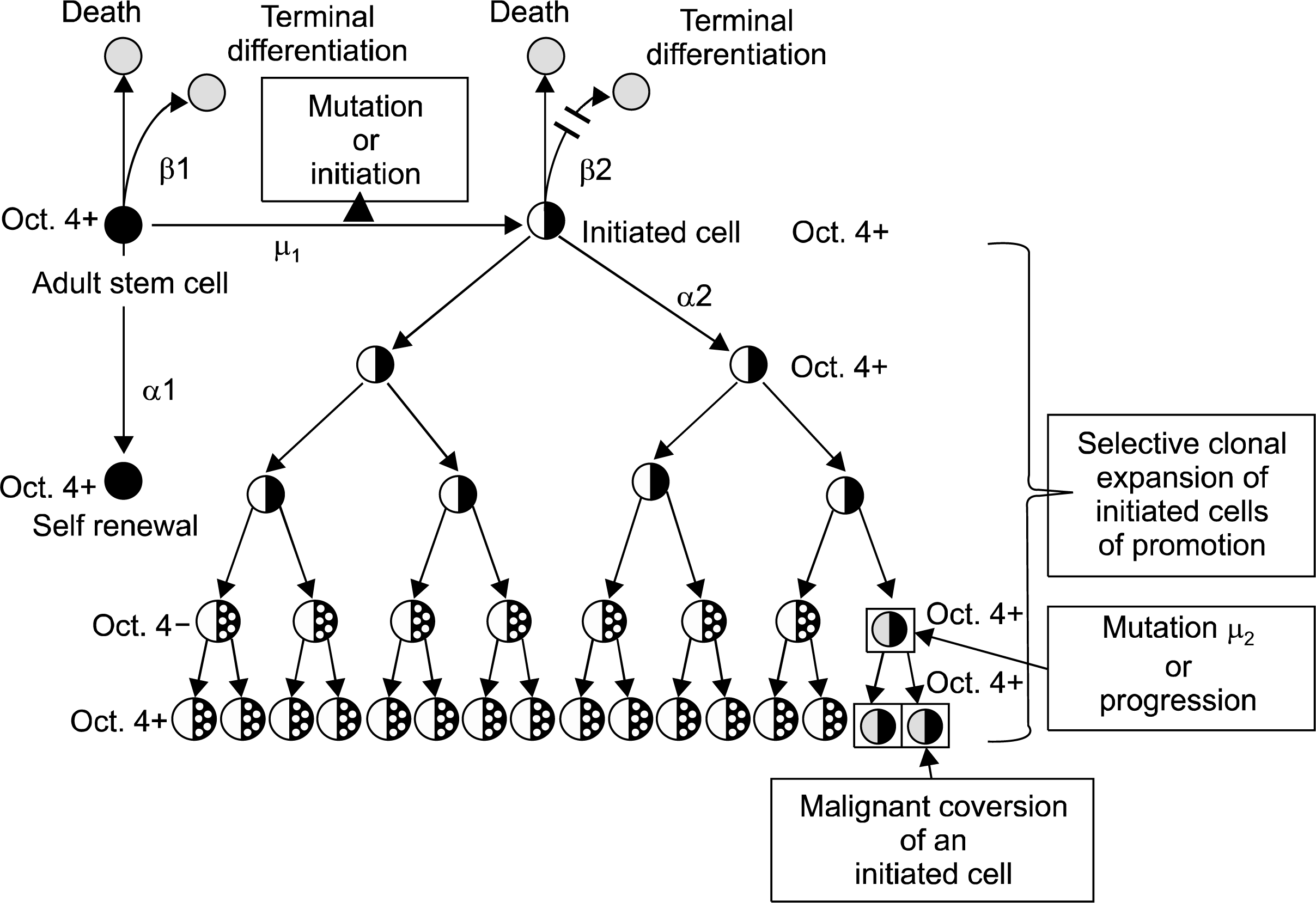
However, just because the organism has initiated cells, it does not automatically mean the organism will get a cancer before death. That single initiated cell must be clonally expanded, during which time, addition changes occur that allow it to acquire the “hallmarks” of cancer (45). This process of promotion helps to define the functional aspects of what “initiation” is. The “immortal” initiated cell, once stimulated to divide, appears to divide only “symmetrically”. Initiation, then, is the process that allows an immortal, stem-like cell to divide only symmetrically. Asymmetrical cell division is, in effect, blocked in the initiated cell under normal conditions.
Promotion is the process by which the single “immortalized” initiated cell can proliferate, symmetrically, to expand the numbers and to not die by apoptosis (46). The combination of increasing cell proliferation without cell death leads to an accumulation of abnormal cells in a tissue. The papilloma in the skin, the enzyme-altered foci of the liver, the nodule of the breast and the polyp of the colon are examples of these expanded initiated cells via the promotion process. Promotion is a process that is caused by stimulating the initiated cell to proliferate by releasing the initiated cell from surrounding mitogenic-suppressing effects mediated by gap junctional intercellular communication (GJIC) of contiguous neighboring cells (47) or by secreted negative growth factors (48). Unlike the initiating event that can occur almost instantaneously in a gene (DNA damage, error-prone repair of that damage, and creation of a mutation or a mutation caused by an error of DNA replication), promotion must occur because (a) the threshold level of mitogenic signaling by non-mutagenic chemicals, growth factors, cytokines, by inflammatory agents (bacterial, viral, fungal, solid particles) or by compensatory hyperplasia caused by cell removal or cell necrotic death.; (b) exposure occurs over a regular interval; (c) occurs for a long period of time and (d) occurs in the absence of anti-promoters (49).
Most, if not all, tumor promoting conditions are associated with the reversible down regulation of GJIC (37–50). The reversible down regulation of exogenous tumor promoters explains the observation that phorbol ester promoted papillomas of mouse skin can regress if the promotion application of phorbol ester is stopped (51) or if agents can prevent the down regulation of GJIC by the promoter, such as chemopreventive chemicals such as green tea components, resveratrol, etc. (49).
Progression is the final operational step that allows a single initiated cell to escape, stably, the dependence on an external (i.e., phorbol ester) or endogenous (i.e., estrogen) tumor promoter for expansion. At this point, all the many genotypic and phenotypic changes needed for the initiated cell to invade, with its own growth stimulating factors acting as its endogenous promoter, the original tissue and to metastasize to a distal site.
Interestingly, oncogenes, such as ras, neu, mos, raf, src, can stably down regulate GJIC (37). Therefore, the progression phase of carcinogenesis might be when the GJIC or the mitogenic suppression by a negative secreted factor of an initiated cell is stably down-regulated.
Now to examine the question, “What is the nature of that single cell that can be initiated to start the multi-stage, multi-mechanism process of carcinogenesis?” As stated earlier, the prevailing idea was that a normal, mortal, differentiated cell could be “immortalized” during the initiated step. This concept has been supported by the observation that, first, an immortal cell could be isolated from a primary rodent cell population with the transfection of the myc oncogene. Once immortalized, transfection of these immortalized cells with the ras oncogene led to neoplastically-transformed cells (45). Implicit in the paradigm, based on this experiment and its interpretation, “de-differentiation” or “re-programming” of differentiated somatic cells occurred when the myc oncogene was integrated and expressed. Only when the cells were “immortalized” could they escape senesce and proliferate indefinitely to accrue all the other changes need for acquiring the “hallmarks” of cancer.
However, as will be discussed in more detail later, there is another interpretation. The new interpretation challenges this mortalization to immortalization process of initiation (8). The alternative hypothesis refers to the older idea that the single cell in the adult tissue that is initiated is the normal, immortal adult stem cell. In this hypothesis, adult stem cells, which exist in most, if not all, adult tissues, are the target cells for initiation. If the initiated gene somehow blocks the normal, immortal adult stem cell from terminal differentiation (i.e. blocks its ability to divide asymmetrically), it will still be able to divide symmetrically to produce two daughter initiated cells that cannot differentiate or apoptose under normal conditions. As a result this initiated adult stem cell, upon mitotic stimulation, will accumulate, as it can not terminally different, senesce or apoptose. Each time it is stimulated to divide, it has the chance to produce more genetic and epigenetic changes needed to become a metastatic cell (Fig. 2).
iPS cells: mortality to immortality?
An amazing series of reports have appeared in the last few months, based on the assumption that reprogramming of genes in a differentiate cell could be accomplished by restoring the activity of a small number of genes thought to be responsible for maintaining “stemness” of embryonic stem cells (52–71). Included in these first attempts were the genes, Oct4, Sox2, c-Myc, and Klf (51). The approach was to integrate these exogenous “stemness” genes into adult differentiated skin fibroblast cells. With successful isolation of mouse, monkey and human embryonic-like stem cells, using several variations of this approach, the emergence of the iPS (induced pluripotent stem) cells opened up a new potential approach to circumvent the ethical, political and legal issues related to isolating human embryonic stem cells (72), and overcoming a few of the serious medical issues related to stem cell therapy. The major problem that could be overcome is the immune rejection of any iPS that might be isolated, manipulated ex vivo and used for therapeutic replacement of tissues of the donor individual, now to be the recipient of his/her own iPS-derived cells.
However, the technique used in these early iPS studies still carry greave limitations for their use, the first is that the definition of an embryonic stem cell or iPS-embryonic-like stem cells is a functionality test, namely these cells must produce teratomas when placed back into an adult animal. In addition, genetically engineering the primary differentiated cells, either with transfection techniques or with the use of gene-carrying viruses, induces the possibility of insertional mutagenesis, together with gene imbalances which might alter gene expression. To use these iPS cells, ex vivo, to produce specific cell types, i.e., specific neurons, insulin-producing beta-cells, osteoblasts, etc., has its own difficulties, in that, “Are these iPS-embryonic-like stem cells capable in vitro to accurately re-capitulate the exact gene patterns in the total genome that reproduces what happens naturally during development of that particular cell type.” Also, to replace the in vitro-iPS-derived differentiated cells, when replaced back into the adult patient, would, in most cases, have to be repeated because the differentiated cell would eventually die. If the pool of iPS-derived cells contained a few iPS-embryonic-like stem cell, there is, then, a possibility of a teratoma being formed. If future techniques can eliminate these iPS-embryonic-like stem cells from forming teratomas when placed in the adult micro-environment, there might even be the chance that these cells when placed into the organ needing tissue replacement, can be “re-programmed” by the specific micro-environment to differentiate into the correct cell type. If this iPS procedure will replace all other embryonic stem cell approaches, not only over coming its own therapeutic limitations be important, but understanding the basic mechanisms of “re-programming” the molecular events will be required in order to regulate specific genes out of the total genome that determine the specific cell type needed for the therapy.
There is, however, another potential explanation of the successful isolation of iPS cells. While it is very obvious isolation of these iPS can be done, challenges to the interpretation of these studies has been reported (72–74). Fundamentally, these reported challenges to the interpretation that the iPS cells were “re-programmed” adult somatic differentiated cells pointed out that the true origin of these iPS cells was not determined. This point was raised in a short analysis of these reports of iPS (75).
An alternative view of the origin of these iPS cells, which is found in the Trosko (8) report came from research on the stem cell theory of cancer. Probably the strongest evidence in support of the adult stem cell as the target cell to initiate the carcinogenesis process comes from the study of leukemia (76). However, another series of cancer studies on solid tumors comes from observations not cited by the stem cell biologists. They started with the old observations that one of the general distinctions between normal cells and cancer cells was the lack of “contact-inhibition” (77) and gap junctional intercellular communication (GJIC) in cancer cells (78). Later, studies linking gap junctions with differentiation and development (33) were made. However, an unlikely source of the critical observation linking stem cells with neoplastic transformation came from the experiments designed to understand the inconsistent results, using embryonic Syrian hamster cells, to test carcinogens (79). The final resolution of this problem, which is rarely cited, was that if the population of embryonic Syrian hamster cells did not include “contact-insensitive” cells, no matter how hard one tried to neoplastically transform this population, no transformants were recovered. Our lab, which was studying gap junctional intercellular communication at the time, as well as being believers in the stem cell theory of carcinogenesis, assumed that these normal “contact-insensitive” cells were adult stem cells in the primary culture.
With this idea in mind, we felt in human beings, adult stem cells must exist in all organs from which tumors arise. We then designed a “kiss of death” assay to detect and isolate these presumptive adult stem cells (80). In brief, we assumed all normal tissues would contain at least three types of cells, namely, the few stem cells; the many transit-amplifying or progenitor cells and the terminally-differentiated cells. Given the knowledge at that time, we knew that the progenitor cells and many terminally differentiate cells express gap junctions at least in some time of their differentiation, we felt that the adult stem cells, in a tissue, surrounded by their GJIC-competent cells, had to be sequestered from their progenitor/differentiated daughters either by some physical barrier or by the lack of functional gap junctions. Clearly, if an adult stem cell communicated directly via gap junctions with their differentiated progeny, they would lose their primitive gene expression identification of “stemness”. Our “kiss of death” assay was based on using a lethally-irradiated human fibroblast layer (these fibroblasts communicated with each other even in this condition). On this “feeder-layer”, we placed disassociated cells from normal kidney tissue. The progenitor cells coupled with the dying fibroblast and in a few days they died (did not proliferate). The terminally differentiated cells, by definition, also, did not proliferate. However, after a week, small colonies of cells appeared, which were shown later to be absent of GJIC and have the ability to differentiate and have a key marker for “stemness”, namely, Oct-A gene expression (81, 82). As a control, we tested 10 human carcinoma cells, which were known not to have functional GJIC. All of these cancer cells grow into colonies. Later, a number of other human epithelial adult stem cells were reported to lack functional GJIC (83).
However, these normal stem cells were shown, not only to meet the definition of what a stem cell should be (have self-renewal and differentiation potential), but also that they could be neoplastically transformed. Historically, one must remember, only of few reports of the neoplastic transformation of human fibroblast and epithelial cells have been made.
The origin of the few that were claimed to have been neoplastically transformed were not identified as using “immortalizing viruses”, such as SV40 or human papilloma viruses or being transfected with hTERT.
In these reports, investigators recovered “immortalized” human cells from normal primary cultures of adult tissues (84–87). Many of these immortalized” cells could then be neoplastically transformed. All of this supports, on the surface, the idea that these normal human primary cell populations could be “re-programmed”. Moreover, it tended to support the hypothesis that one must first “immortalize” a normal “mortal” cell in the primary culture before it could be neoplastically transformed (44).
Alternatively, since the normal adult cell is “immortal” until it is induced to “mortalize” or terminally differentiate, apoptose or senesce, it will remain immortal. If a virus, such as the SV40, human papilloma, or human hepatitis, blocks the immortal normal adult stem cell from terminally differentiating, apoptosing or senescing, it will remain immortal. It does not become “reprogrammed” to the stem cell state. In fact, it has now been blocked from normal programming of differentiation, apoptosing or senescing.
In a series of reports on the isolation and partial characterization of human breast stem cells, it was shown that these cells could differentiate into human breast epithelial cells, form true-like mammary structures, transit from a completely mutually exclusive set of genes in the stem cell state to another set in the differentiated state (88). Later after exposure to SV40, clones were obtained that were “immortalized” or more accurately, blocked from “mortalization”, which maintained most of the marker genes of the normal stem cells. After X radiation, a few clones were obtained that, again, maintained these critical stem cell marker genes and were weakly tumorigenic. After transfection of these weakly tumorigenic cells with the neu oncogene, clones of highly tumorigenic cells were isolated (89). Again, these cells maintained the critical set of expressed genes found in the original normal adult human breast stem cells (89). Much later, these, and other normal human adult stem cells were shown to express the Oct-4A gene and not express the connexin genes or have functional GJIC (83). In summary, these results demonstrate, directly, that the Oct-4A gene was not “re-programmed” during this process of becoming “immortalized” and of becoming neoplastically transformed.
One addition implication of these findings is that the claim that viruses can cause cancer might have a logical foundation in these observations. If after exposure to normal human organs, which contain adult stem cells, any virus that might infect an adult stem cell might prevent that stem cell from differentiating, apoptosing or senescing. In effect the virus has “initiated” this adult stem cell. As a result, this cell could live long enough to accrue addition changes to become a malignant cell.
An addition significant observation has been made with regard to the interpretation of iPS cells being the result of “re-programming” of differentiated cells. In the study where mouse iPS cells were isolated from primary cultures of mouse heptocytes, a comment was made in the paper (71), namely: “The mechanism of iPS cell induction, however, is unknown. Low efficiency of iPS cell induction suggests that their origins may be of undifferentiated stem cells co-existing in fibroblast culture. ...Most of them were also positive for B-gal, indicating that iPS-Hep cells were derived from hepatocytes or other albulmin-expressing cells, but not from un-differentiated cells that do not express albumin.” However, in the isolation of human liver stem cells (90), the liver stem cell, an undifferentiated cell, by definition, expresses, not only Oct-4, but also the albumin gene and protein. This further provides evidence that the origin of these iPS cells was not the “re-programming” of differentiate hepatocytes but the selection of the adult liver stem cells.
Adult stem cells, cancer stem cells and cancer non-stem cells
If the aforementioned hypothesis is correct, namely, that the adult stem cell is the target cell for initiating the carcinogenic process, then one might be able to test part of this hypothesis by looking a one of the markers for these adult stem cells, namely, Oct4A in spontaneous tumors. To begin, it is already known that all tumors lack functional GJIC, either because they never express their connexin genes or that the expressed connexins are rendered non-functional by expressed oncogenes (48). In the case of initiation/promotion models, such as the rat liver studies (42), the lesions appear to express connexins and have functional GJIC, until exposed to tumor promoting chemicals, such as penobarbital, known to reversibly inhibit GJIC (91–94). On the other hand, there are tumor cells that do not express any connexins (95, 96). Yet, when cell lines are derived from tumors and growth in vitro, the micro-environment changes, as it does in vivo.
Even in the case of Hela cells, cells that, when cultured, under certain conditions, have no expressed connexins or functional gap junctions. However when grown under another condition, such as co-cultured with HL-60 leukemic cells or with normal fibroblasts, both cell types can be shown to be coupled by gap junctions (97, 98). Even when one examines the promoted lesions of a rat liver, one can detect “clones within clones”, or in general, there are sub-populations within all tumors. The interactions, both by direct contact and by secreted factors, influence the growth of the whole tumor (99). This indicates that, as an initiated cell is promoted, soon, additional genetic/epigenetic changes occur due to micro-environmental changes. Eventually, a single initiated cell accrues all the hallmarks need to invade and metastatize. During that process, the original adult stem cell, when initiated (blocked from asymmetric cell division or terminally differentiation), starts to grow. As the clone of initiated cells growths, the micro-environment changes between the initiated cells and the neighboring normal cells [There will be stromal-epithelial interactions (100–102), as well as interactions between the initiated cells and themselves in the interior of the tumor (103)]. The induced intracellular signaling, caused by these micro-environmental changes, are bound to alter gene expression (104).
The altered gene expression in these initiated cells could induce some genes that could induce apoptosis, partial differentiation or senescence in these cells. These cells might be the so-called “cancer non-stem cells”.
Given the hypothesis that the normal adult stem cell is the target cell for initiation, and that initiation blocks the stem cell from terminal differentiation, it was reasonable to expect that tumors contain “cancer stem cells”. These would be the cells that helped to sustain the long term growth of a tumor. When cells were isolated from tumors that were either able or not to perpetuate the tumor (105, 106), a large number of “cancer stem cells” from many different types of tumors have been and are continuing to grow (107–122). With the demonstration that Oct-4 gene was considered a stem cell marker (83), not shown to be found in normal tissue, but found in various tumors (123–125), it was hypothesized that this Oct-4 gene was re-expressed during the carcinogenic process. This reflected the prevailing paradigm that a normal differentiated somatic cell had to be “reprogrammed”. However, since our laboratory had isolated many human adult stem cells from various tissues (kidney, breast, pancreas, mesenchyme, liver, intestine) (80, 126–132), we were able to demonstrate that all of these normal adult organ-specific stem cells expressed Oct-4A and did not express connexin genes or have functional GJIC (32, 83).
Moreover, in tumors from these organ sites, all expressed Oct-4A within the tumor and in cancer cell lines, such as HeLa and MCF-7 (8, 83). Oct-4 has also been shown in other cancer cell lines (133). This, in fact, suggests that primary cell lines, that eventually senesce or go through “crises”, do so because the few stem cells have been diluted out during subsequent passages and that the progenitor cells exhibit the “Hayflick phenomenon” (5). Immortal and cancer cell lines must, by definition, include stem cells that have been initiated or initiated and neoplastically converted to become cancer stem cells.
In both cell culture and within the tumor, one would expect the micro-environment to change so as to induce, differentiatially, altered gene expression causing some of these Oct-4 cells positive cells to repress its expression. This would cause these cells to eventually cease to proliferate indefinitely.
Upon testing the hypothesis that the Oct-4A positive cells within a tumor were the “cancer stem cells”, it was shown that Oct-4A positive cells were found in 100% of the 21 canine different tumor sites (123). Later, it was shown to be expressed in most of the human bladder tumors examined for Oct4 (124). Still later, Oct-4 was observed in human oral cancers (125). What was also shown in all these tumors was the ratio of Oct4 positive cells to Oct-4 negative cells varied widely. Even when one examined the cancer cell lines, HeLa and MCF-7 cells, the population of cells exhibited a mixture of Oct-4 positive to Oct4-negative cells. The implications of this for both molecular/biochemical studies of cell lines derived from tumors or from the tumors themselves is that one needs to be cautious of the results because these cell line and tumors are not homogeneous. That especially relates to many DNA micro-array studies which would be the net-effect of a mixture of cancer-stem cells and cancer non-stem cells.
Cell-cell communication and differentiation/growth control/apoptosis: role in chemoprevention and chemotherapy
If one starts with the premise that the adult stem cell is the target cell that starts the initiation/promotion/progression process of the multi-stage, multi-mechanism theory of carcinogenesis, strategies for the prevention and treatment of cancer seem fairly obvious. First, simply by increasing or decreasing the stem cell pool in any organ would, all other factors being equal after initiation, increase or decrease the probability of the initiation event from taking place. While it would be appropriate to minimize exposures to mutagens, such as protect oneself from too much sun-exposure, it would decrease the risk to sun-induced skin cancer. However, it is impossible to reduce to zero the risk to the initiation event. Every time a cell proliferates, there is a finite chance that an error in replication could lead to a mutation in a critical gene involved in the carcinogenic process.
That, then, leads to the importance of preventing the initiated stem cell from being promoted (by preventing these initiated cells from being expanded and/or being induced to die by programmed cell death or apoptosis). Since both normal adult stem cells and their initiated progeny are growth controlled either by secreted negative growth regulators or by gap junctional intercellular communication, and since most, if not all, tumor promoting agents block cell-cell communication between the initiated cell and the normal differentiated or neighboring sib cells (37–40), it would seem that the tumor promotion phase would be the most effective phase to intervene. By interfering with the tumor promotion phase, which, in the case of human beings, can take place over decades, would allow one to delay or even reverse the clonal expansion of the initiated cells to accrue all the hallmarks of a cancer cell.
Given that endogenous and exogenous factors can act as tumor promoters, and given that tumor promotion, caused by very different conditions (e.g., wounding, normal growth, cell removal, cell death, drugs, pollutants, food additives, toxicants, microbial toxins, hormones, inflammatory factors, solid particles, etc.) (7), it might seem that it would be impossible to prevent tumor promotion. In deed, tumor promoters can be species-, gender-, developmental stage-, cell-type and organ-specific. However, there are some universal characteristics of tumor promoters, namely, they seem to have threshold levels of action; exposure must be for long periods of time, given in regular exposures and in the absence of agents that are considered “anti-promoters”. Also, one of the emerging observations that seems to link two different physiological processes (cell proliferation and the immune system) with the tumor promotion process is chronic inflammation (7).
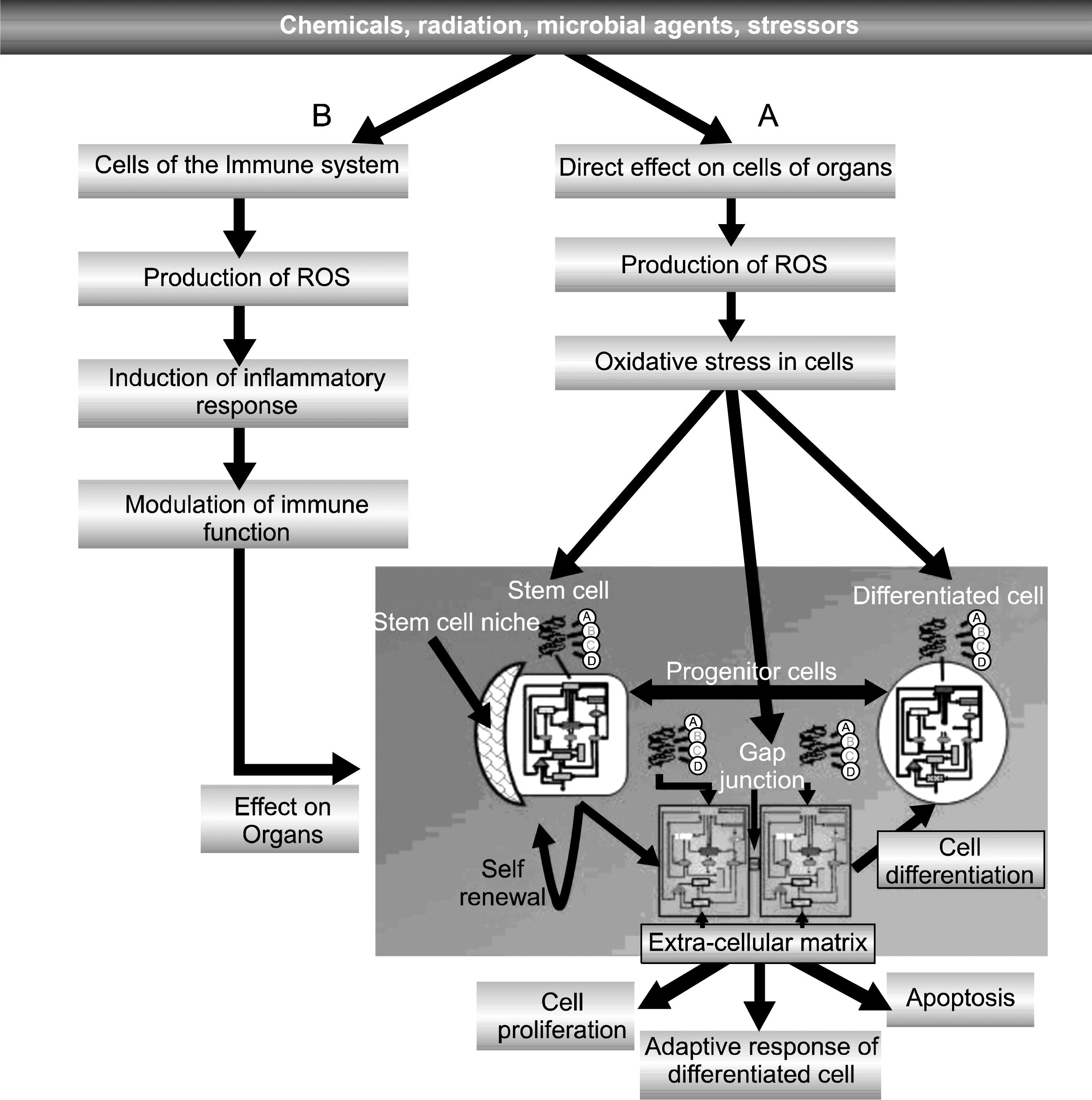
The hypothesis seems to be that when, in an initiated tissue having an adult stem cell that is blocked in its ability to proliferate by anti-mitogenic factors, is exposed to an agent/condition that triggers oxidative stress in both cells of the immune system and the initiated epithelial or fibroblastic tissue, an interaction between the two can happen (Fig. 3).
Tumor promoters appear to be non-genotoxic (134), yet they can induce oxidative stress (135). Contrary to common belief, while radical oxygen species (ROS) can, in principle, damage any macromolecule in a cell, in the target cell at tumor promoting agent at non-cytotoxic levels and in an undifferentiated initiated stem cells, they can induce intra-cellular signaling leading to (a) inhibiting GJIC and (b) altering gene expression. Even a genotoxic agent, such as UV light, by killing a large number of cells in initiated skin, can cause compensory hyperplasia of any surviving initiated cell. Cytotoxic agents, such as the non-genotoxic alcohol or carbon tetrachloride, can also induce compensatory hyperplasia to act as an “indirect tumor promoter”.
The search for a universal chemoprevent agent might now be seen as an illusion. It has been shown that a dietary component, beta-sitosteriol, could prevent the growth of initiated rat liver cells with expressed ras-oncogene, but not the same type of rat liver cell expressing oncogenes, such as neu, src or even myc-ras (136).
In effect, tumor promotion prevents the initiated cell from terminally differentiating and from dying by apoptosis. The accumulated initiated cells expand and increase the probability of additional genetic and epigenetic changes. Therefore, if oxidative stress is a major component of tumor promotion, then anti-promoters or chemo-preventive agents might be viewed by acting by anti-oxidant mechanisms. Evidence exists that many anti-promoters have anti-oxidant properties. However, one must be careful in applying this observation in that some anti-oxidants can have pro-oxidant activity (137). Unless one understands the actual mechanism by which a chemo-preventive agent works, unintended consequences could occur. The classic examples of this were the termination of the CARET and ATBC clinical intervention trials to reduce the risk to cancers (138, 139).
Since caloric restriction has been shown to reduce many chronic diseases (140–142), one might hypothesize that this physiological phenomenon might reduce both the stem cell pools of certain organs, as well as prevent cell proliferation and possibly induce apoptosis of any initiated cells (143). Therefore, caloric restriction might act both to reduce the initiation of stem cells and to inhibit the promotion process. This might explain the relative low incidence of cancer in the survivors of the atomic bombs (143).
In summary, there seems to be an important role in cell-cell communication in regulating, not only cell proliferation, cell differentiation and apoptosis of normal stem and progenitor cells, but also of the initiated cell. Interference of cell-cell communication by all the agents and conditions which are associated with tumor promotion has been documented. Tumor promotion is that process that allows the initiated cell, which can not normally divide by asymmetrical cell division and does not normally apoptose, to clonally expand, allowing addition genetic changes to become a malignant, metastazing cell. Prevention of the down regulation of cell-cell communication by tumor promoters and the restoration of cell-cell communication in tumor cells would be the strategy for chemoprevention and chemotherapy, respectively (49).
Barker hypothesis/nutrition and stem cell biology
To prevent or treat cancers, with the assumption that the adult stem cell is the target cell for initiating the cancer process, the old Barker hypothesis (144, 145) seems to be explained, mechanistically, with the stem cell theory. There are several excellent examples of this.
The pre-natal exposures to the drug, DES, led to increased risk to vaginal cancers of the female offspring of mothers who took the drug during pregnancy (146).
If the DES exposure during pregnancy of a female fetus causes an increase in stem cell pool in the vaginal tissue, then during sexual maturation of the vaginal tissue, any initiated cell caused by abnormal proliferation might now be promoted by the sex hormones at this stage of development.
Another example is the study of the breast cancers in Japanese women who survived the atomic bombs of Hiroshima and Nagasaki (147). One explanation is that Japanese mothers’ diets included lots of soy products and, in part, these women were calorically restricted (142). Human breast stem cells, having been isolated and characterized (88), have been shown to be induced to differentiate into progenitor breast epithelial cells by a major component of soy, namely genistein (148). Only the human breast stem cells, but not the differentiated progenitor breast epithelial cells, were capable of being neoplastically transformed (89). Therefore, if the soy-diet of pregnant Japanese women induced differentiation of the adult breast stem cells of the female fetus, as it has been shown in rodents (149), then after birth, there would be fewer breast stem cells for the development of breast tissue and fewer adult stem cells to be targets for initiating the breast carcinogenesis process.
Recently, in experimental pregnant rats exposed to the environmental pollutant, bisphenol-A, the male offspring after birth developed a high risk to prostate cancers, even though they were not exposed to this pollutant after birth (150). Even more extraordinary was the observation that if the pregnant rats were exposed to both bisphenol-A and dietary soy, the male rats had a dramatically reduced risk to prostate cancers, again even though after birth they were not exposed to either factor. This strongly suggests that the pollutant and dietary factors acted on the target cells for prostate cancer. One possible explanation is that bisphenol A caused an increase in the prostate stem cell pool in the male fetus. This would increase the risk for an initiation event. After birth, some factor, either endogenous or exogenous, could promote the prostate cancer. On the other hand, during development of the male, soy diet, containing antioxidants, such as Bowman-Birk inhibitor (151, 152) or genistein might cause the prostate stem cell to differentiate, reducing the stem cell pool and reducing the risk for prostate stem cell initiation.
Recently, it was reported that there was a correlation of umbilical cord blood haematopoietic stem cell and progenitor cell levels with birth weight. This implied that there might be a prenatal influence on cancer risks (153). Possibly, the fact that the frequencies of childhood cancer types, which are, in general, different from the types of adult cancers, might also be related to this Barker hypothesis. In general, cancer is usually viewed as an “old-age” disease. Therefore, while cancers in children are rare compared to adult cancers, their types seem to be different than the adult cancers. From the teratomas to the other neuronal and lympho-reticular tumors, one might have characterized them as primitive-like. In addition, today, the “success-rate” of treating childhood cancers seems much better than the success rate of adult cancers.
If during early development, the stem cells of the origin of childhood cancers are increased, initiation is increased due to enhanced errors of replication, or possibly, due to epigenetic alteration (154), not mutation of oncogenes/tumor suppressor genes, then exposures, postnatally to massive amounts of normal growth factors of childhood, could lead to the “promotion” of these stem cells. These tumors, if they are of non-irreversibly altered stem cells, are now exposed to agents that can cause them to terminally differentiate, they might be treated easier than the tumor cells of adults which were irreversibly initiated or mutated and then promoted over decades to accrue many more alterations in their genome to become an “cancer stem cell”.
In summary, based on the assumption that (a) the stem cell pool in specific tissues can be modulated (increased or decreased) during development of the fetus, and (b) that the stem cell is the target cell for initiating the cancer process, dietary or pollutant/drug exposure of the fetus could dramatically increase or decrease the risk to cancer later in life. This could be the explanation of the Barker hypothesis. Therefore, implications for prenatal care of pregnant women should be of high priority. It is this fact that takes one’s control of cancer risk out of the individual’s hands. Only after one can, in part, control one’s own behavior, can one have partial control to prevent the promotion of ones initiated cancer stem cells (which all human beings have in their bodies).
Stem cells and aging
With the recent focus on stem cell biology, stem cells as a target for diseases and stem cell therapy, it would seem that its possible role in the aging and diseases of aging would have been a predominant component of the theories of aging. However, only within the recent decade, have speculations that stem cells must play a role in the aging process appeared (155–159).
The classic dilemma in the aging field involved the issue that the aging and diseases of aging, such as cancer, are or are not independent processes. When one examines many genetic predispositions to cancer, such as xerodermaq pigmentosum or Downs, predisposition to aging also occurs in the skin, where ultraviolet the environmental trigger both skin cancer and aging of the skin, and predisposition to leukemia and high risk to Alzheimer’s, respectively (23, 160).
Exogenous agents that can induce cancers could also induce premature aging. In natural aging, we notice that the individual organs do not uniformly age. The individual that is an alcoholic is at high risk for liver cancer and “aging” of the liver function. The young persons, who expose themselves to large amounts of sun light, will increase the risk for skin cancer and pre-mature aging of the skin. A cigarette smoker induces pre-mature aging of lung function, while at the same time enhances the risk for lung diseases, such as lung cancer.
In deed, even though one might note that many chronic diseases appear was we chronically age, the fact that cancers appear in young children seems to challenge the idea that the aging process is causally linked to the carcinogenic process. However, it should be noted that the types of tumors in children appear to be very different than those found in adults, in that they appear very “primitive-like”. To date the success frequency of cancer therapy for childhood cancers is significantly better than most adult-type tumors.
Factors associated with both the diseases of aging, oxidative stress and inflammation, as well as factors, such as anti-oxidants and caloric restriction, seem to be correlated with the reduced risk to aging and chronic diseases. In brief, In brief, the pre-mature or progressive loss of structure/function of cells/tissues/organs/organ systems is a common feature of aging and disease. Depending on the circumstances by which genetic and environmental/dietary agents interact, either or both the loss of efficiency in biological function (aging) or an appearance of clinical abnormality (disease) can occur.
If the hypothesis presented here is correct, namely, that the adult stem is the target cell for cancer and if the stem cell pool (increased or decreased) alters the risk to cancer, then a possible linkage can be made where the aging process and the cancer process (as a model for other stem-cell-dependent chronic diseases) share a common element, namely the adult stem cell. Since biologically the terminally differentiated cells must be derived from the transit-amplifying or progenitor cells and the adult stem cells, it should logically follow that reducing the stem cell pool would reduced the risk of cancer and be linked to the inability to expand tissue and repair.
The amazing recent discoveries of two genetic syndromes, namely the Hutchinson Gilford-progeria, pre-mature aging syndrome (161–163), and the Nieman-Pick type C neurological syndrome (164), have identified adult stem cells as being the center of both diseases. In the former, a mutated lamin A gene appears to cause abnormal functioning of all the adult stem cells of all organs. On the other hand, the organ-specific neuronal adult stem cell of the Nieman-Pick type C seems unable to differentiate properly in the brain. Even more dramatically, in both cases, exogenous agents seem to be able to circumvent the genetic dysfunction in the stem cells of both diseases (165, 166).
There have been speculations that relationship between aging and cancer were the result of evolutionary forces (167). In essence, the consequences of unlimited cell proliferation would only enhance genomic instability, therefore, senescence was a option a cell had to decrease the risk to cancer (168, 169). Although it is generally assumed that a stem cell is immortal until it is induced to terminally differentiate, no one has yet demonstrated that a single embryonic or adult stem cell can proliferate without senescing at some point [remember, all current attempts to grow embryonic or adult stem cells in vitro are not done under the in vivo niche conditions, it has been shown that hypoxic and anti-oxidant conditions in vitro appear to prevent differentiation or early senesce of stem cells (10)]. While progenitor cells, such as human fibroblasts or epithelial cells have limited life spans in vitro under contemporary conditions., the “Hayflick phenomenon” (4), then the shortening of telomeres, and genomic instability might be characteristics of only the progenitor or transit-amplifying cells.
In summary, conceptually, if adult stem cells can be targets for mutagens, cytotoxic or epigenetic agents that can alter gene expression in a tissue, depending on the gene and the number of stem cells affected, then a chronic disease might manifest itself (diabetes, cancer, atherosclerotic plaque, cataract). On the other hand, if the stem cell pool is increased or decreased during pre-natal/postnatal development, altered risk to chronic disease in later life would be seen. By decreasing the stem cell pool in specific organs or in all organs later in life will hamper the functioning of that organ or individual.
 XML Download
XML Download