

 PDF
PDF Citation
Citation Print
Print


Introduction
Stem cells are characterized by their ability to self-renew and to differentiate along multiple lineage pathways. Therefore, they have significant potential in tissue engineering and regenerative medicine. Since the 1970s, mesenchymal stem cells (MSCs) have received growing interests, attracting an increasing number of people to study their extensive self-renewal, differentiation potential (1) as well as their immunoregulation capability (2-7). Though MSCs were first isolated and cultured from the bone marrow (8), they have since been harvested from many other organs and tissues, including adipose tissue (9), umbilical cord (10), placenta (11), skin (12), muscle tissue (13), neuronal tissue (14), and dental pulp (15). While MSCs derived from bone marrow (BM-MSCs) continue to be the research hotspot, adipose tissue yields about 500-fold greater MSCs than bone marrow with the same quality (16). In addition, adipose-derived stem cells (ADSCs) can be easily isolated from a number of adipose tissues and then expand in abundance ex vivo. Also, ADSCs can differentiate into adipocytes, myocytes, chondrocytes, and osteoblasts (9), and share similar surface markers with BM-MSCs (17). What’s more, ADSCs do not lead to adverse side effects, such as tumorigenicity, chromosomal abnormalities, or immune rejection (18). Aforementioned characteristics make ADSCs attractive candidate for use in clinical therapy and fundamental research.
Two types of adipose tissue are known in mammals, namely white adipose tissue (WAT) and brown adipose tissue (BAT). WAT is widely distributed in subcutaneous tissues and visceral fat surrounding organs throughout the body. WAT’s function is to store the excess energy into form of fat. In contrast, BAT is only observed in limited parts of body, such as interscapular, axillary, and perirenal region, and it disappears from most of these areas with age. BAT is specialized in energy expenditure, and it plays an important role in maintaining body temperature and energy balance (19). However, the differences between ADSCs from BAT (B-ADSCs) and ADSCs from WAT (W-ADSCs) are not well known. Only a few literatures revealed the differentiation of B-ADSCs and W-ADSCs (20), but the difference of their immunoregulation capacity is still unclear. Especially, it’s still unknown whether the capacities of B-ADSCs and W-ADSCs will change as age increases. Therefore, in this study, we provide a general comparison between B-ADSCs and W-ADSCs from both young and aged mice in their isolation and expansion, proliferation and differentiation, and immunoregulation capacities. The comparative study intends to provide the basis for the choice of ADSCs in clinical application and fundamental research.
Materials and Methods
Animals
Healthy C57BL/6 mice (3∼4 weeks and 7∼8 months) were obtained from the Laboratory Animal Center of the Academy of Military Medical Sciences of China (Beijing). All experiments were performed in accordance with the Academy of Military Medical Sciences Guide for Laboratory Animals. Additional approval was granted by the Animal Care and Use Committee of Beijing Institute of Basic Medical Sciences, with the approval number BMS-12021103.
Isolation and expansion of B-ADSCs and W-ADSCs
C57BL/6 mice (3∼4 weeks and 7∼8 months) were euthanized by cervical dislocation and sterilized in 75% ethanol for 5 minutes. B-ADSCs were isolated from interscapular adipose tissue with dorsal side facing up, and W-ADSCs were obtained from inguinal subcutaneous adipose tissue with abdomen facing up (21). Then tissue was rinsed with phosphate buffered saline (PBS), minced, and followed by digestion with 1 mg/ml collagenase type IV (Sigma-Aldrich) and 1 mg/ml dispase (Sigma-Aldrich) for 35 minutes at 37℃ with agitation. To stop digestion, cells were added to α-MEM medium (Gibco, Carlsbad, CA) - containing 10% FBS (Gibco) - and then filtered through a 40 μm cell strainer (Biologix, USA) to generate single-cell suspensions. After centrifugation at 400 g for 5 minutes, cells were re-suspended in α-MEM - containing 10% FBS - at 37℃ in 5% CO2. When they reached 80∼90% confluence, cells were sub-cultured in a ratio of 1:3.
Proliferation and differentiation of ADSCs
For cell proliferation, cells were seeded in a 24-well plate at the density of 1×104 cells per well in cultured medium. Cells were counted every two days for 8 days. The mean cell number was calculated for each triplicate. For differentiation, passage 3 (P3) cells were seeded in a 24-well plate at the density of 1×104 cells per well overnight. They were then induced with two media: one osteogenic induction medium, which contains 0.1 mM dexamethasome, 50 mM ascorbate-2 phosphate, 10 mM glycerophosphate (Sigma-Aldrich); and the other adipogenic induction medium, which contains 1 mM dexamethasone, 200 mM indomethacin, 0.5 mM 3-isobutyl-1-methyl-xanthine, and 10 μg/ml insulin (Sigma-Aldrich). In vitro osteogenesis, cells were cultured for 0, 3, 7, 10 days, and then stained for alkaline phosphatase (ALP) activity, using the alkaline phosphatase kit (Sigma-Aldrich). In vitro adipogenesis, cells were cultured for 0, 3, 7, 10 days, and then stained for fat droplets, using the Oil-Red-O (Sigma-Aldrich). In addition, P1 cells were seeded in a 24-well plate at the density of 1×104 cells per well on carbon nanotube (CNT) for 3 days, and were analyzed for the expression of cardiac troponin T (cTnT), GATA4, and Nkx2.5 by qRT-PCR.
Carboxy fluorescein diacetate succinimidyl ester labeling
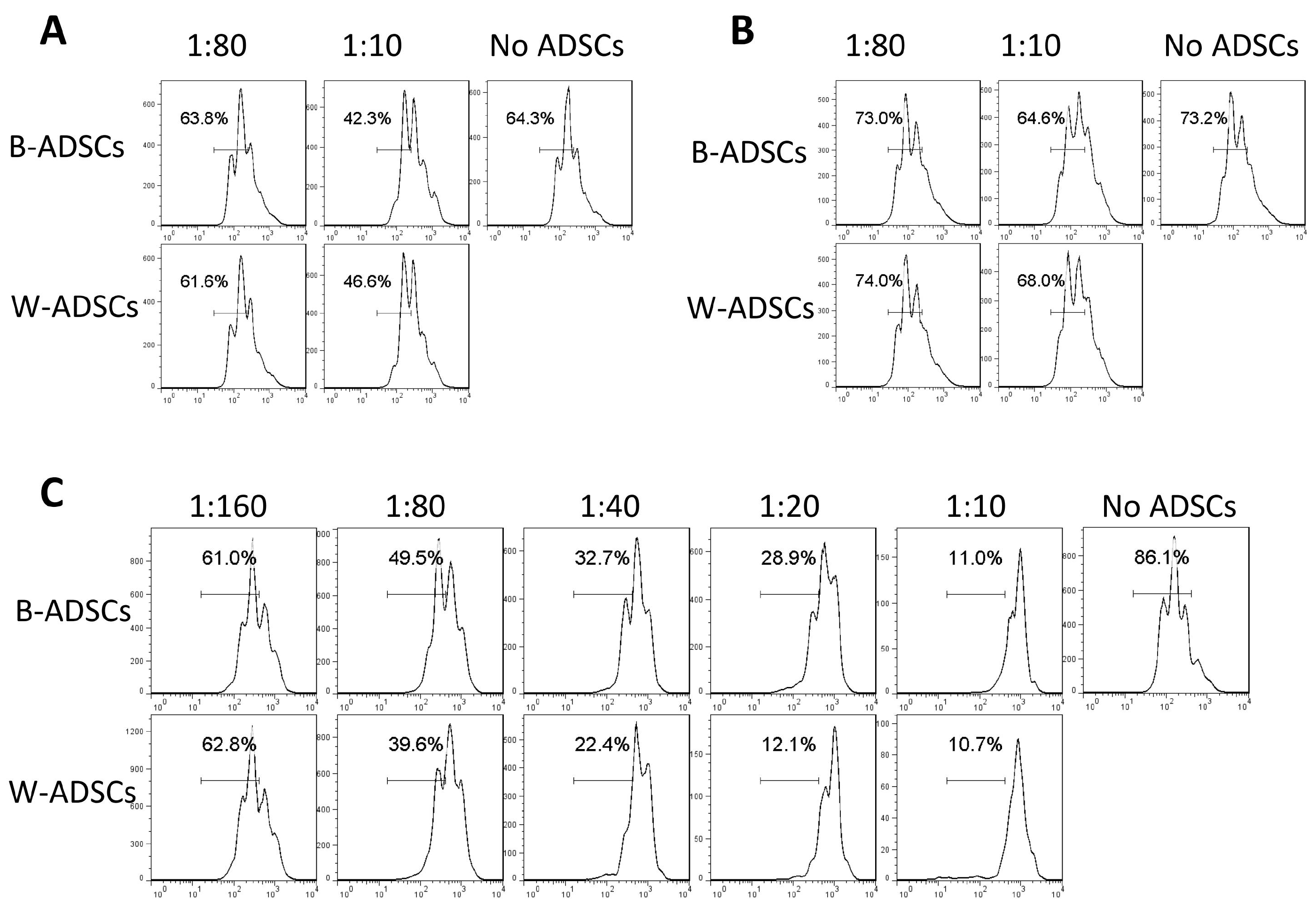
Peripheral CD3+ and CD4+ T cells were selected with CD3e and CD4 MicroBead Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) from spleens of adult C57BL/6 mice, and then labeled with 5 μM carboxy fluorescein diacetate succinimidyl ester (CFSE, Invitrogen, Carlsbad, CA, USA) for 7 minutes at 4℃. Labeling was terminated according to the manufacturer’s protocol. After washing, cells were activated with 50 ng/ml phorbol myristate acetate (PMA) and 1 μg/ml ionomycin (Sigma-Aldrich) for 16 hours, and then co-cultured with or without B-ADSCs and W-ADSCs for 48 hours. Cell division, as indicated by reduction of fluorescence intensity, was analyzed by flow cytometry.
Flow cytometry
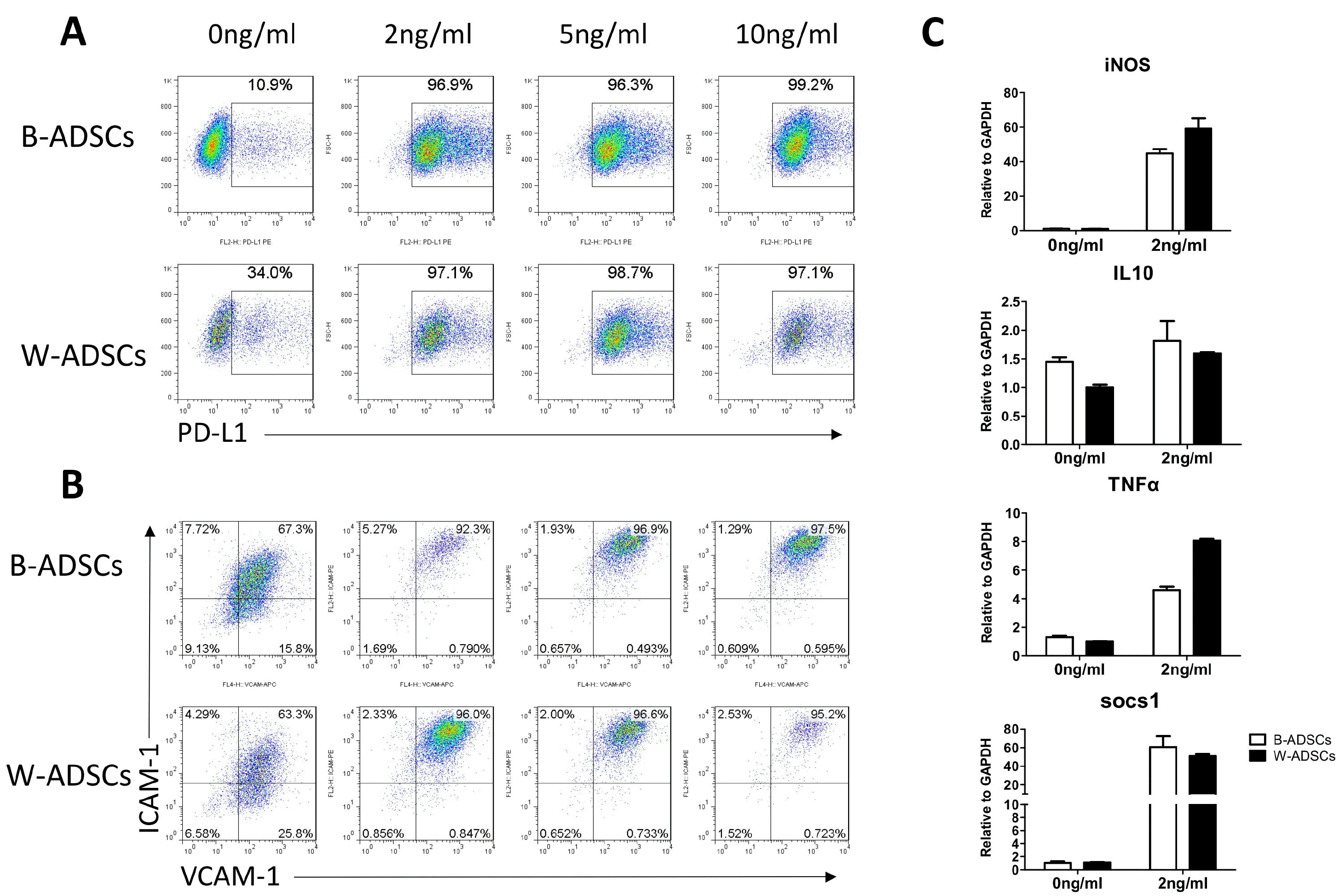
P3 cells (1×105 cells) were suspended in 100 μl PBS and labeled with antibody of surface marker with the following: CD29, CD31, CD45, CD86, Sca-1, and MHC II. Cells cultured with stimulation of 0 ng/ml, 2 ng/ml, 5 ng/ml, 10 ng/ml TNF-α and IFN-γ for 12 hours, and were labeled with antibody of programmed death-ligand 1 (PD-L1), intercellular cell adhesion molecule (ICAM-1), and vascular cell adhesion molecule (VCAM-1) for 30 minutes at 4℃. Cells were then washed with PBS and fixed with 1% paraformaldehyde and detected by using flow cytometry. All antibodies were purchased from BD or eBioscience.
Quantitative RT-PCR
Total RNA was extracted with TRIZOL (Sigma-Aldrich) and reverse transcribed into cDNA with a reverse transcriptase kit (Takara). cDNA was used as a template in real-time PCR with SYBR Green reagent from TOYOBO to determine specific gene expression. Primer sequences were presented in Table 1, and qRT-PCR was performed with the following conditions: 95℃ for 3 minutes; 40 cycles: 95℃ for 15 seconds, 60℃ for 15 seconds, 72℃ for 15 seconds. It was then followed by melting curve analysis.
Statistical analysis
All data were analyzed with Prism 5.0 software (GraphPad Software, CA, USA) and are presented as the mean±S.D. Statistical significance was assessed by unpaired two-tailed Student’s t-tests. p-value less than 0.05 were considered significant. Experiments were performed in triplicate.
Results
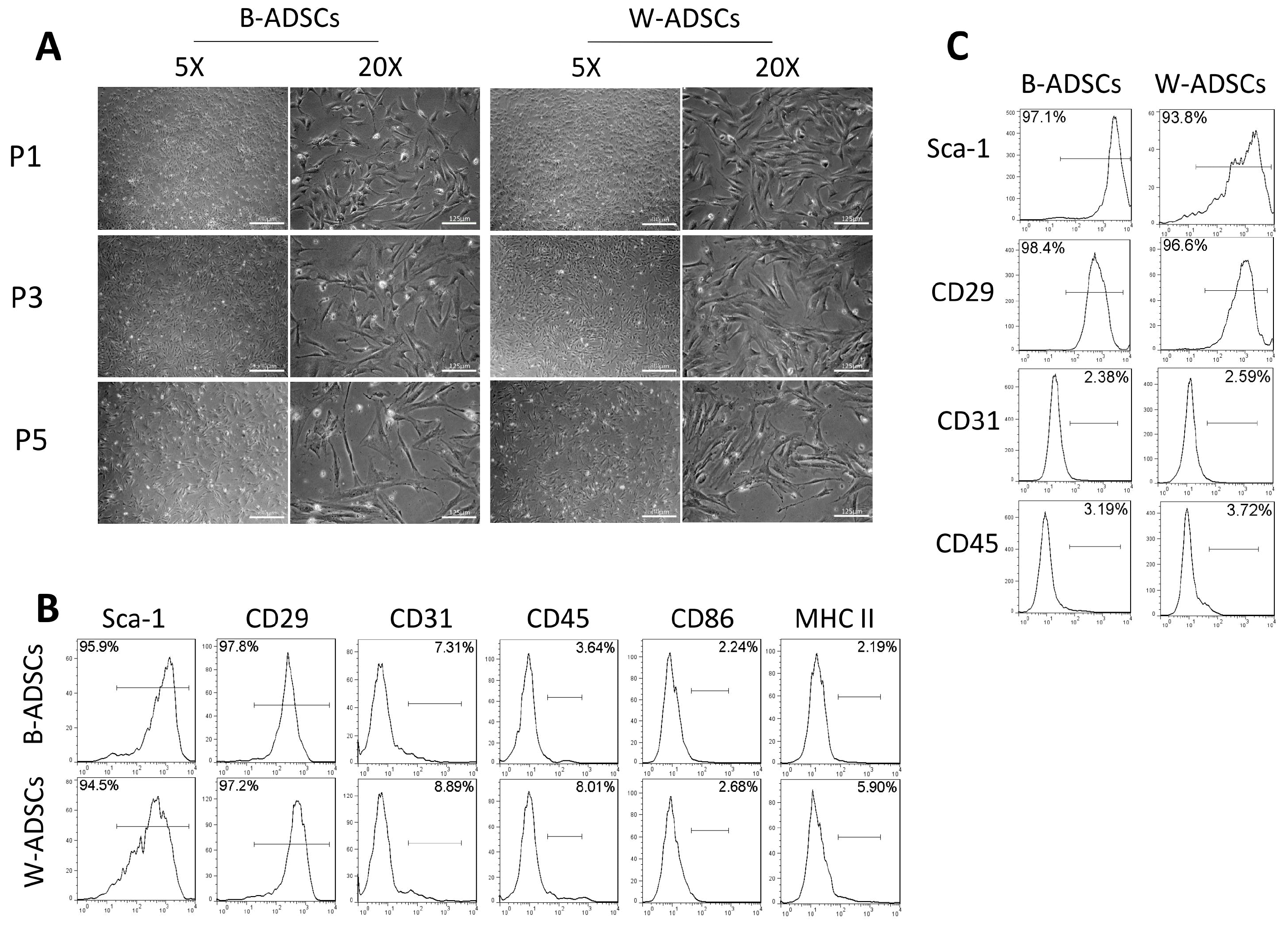
Similar morphological and phenotypic characteristics of B-ADSCs and W-ADSCs
B-ADSCs and W-ADSCs were spindle-shaped and fibroblast-like in appearance 3 days after their isolation, and were spiral-shaped morphology when they reached 80∼90% confluence. After continuous passage, cell morphology of both B-ADSCs and W-ADSCs became larger at P5 (Fig. 1A). These two ADSCs from young mice (3∼4 weeks) were then analyzed for cell surface molecule expression: positive for the MSC markers Sca-1 and CD29, but negative for the haematopoietic and endothelial markers CD45, CD31, and MHC class II (Fig. 1B). Meanwhile, B-ADSCs and W-ADSCs from aged mice (7∼8 months) expressed the same cell surface marker (Fig. 1C). These results indicate that B-ADSCs and W-ADSCs exhibited similar morphology and immunophenotype.
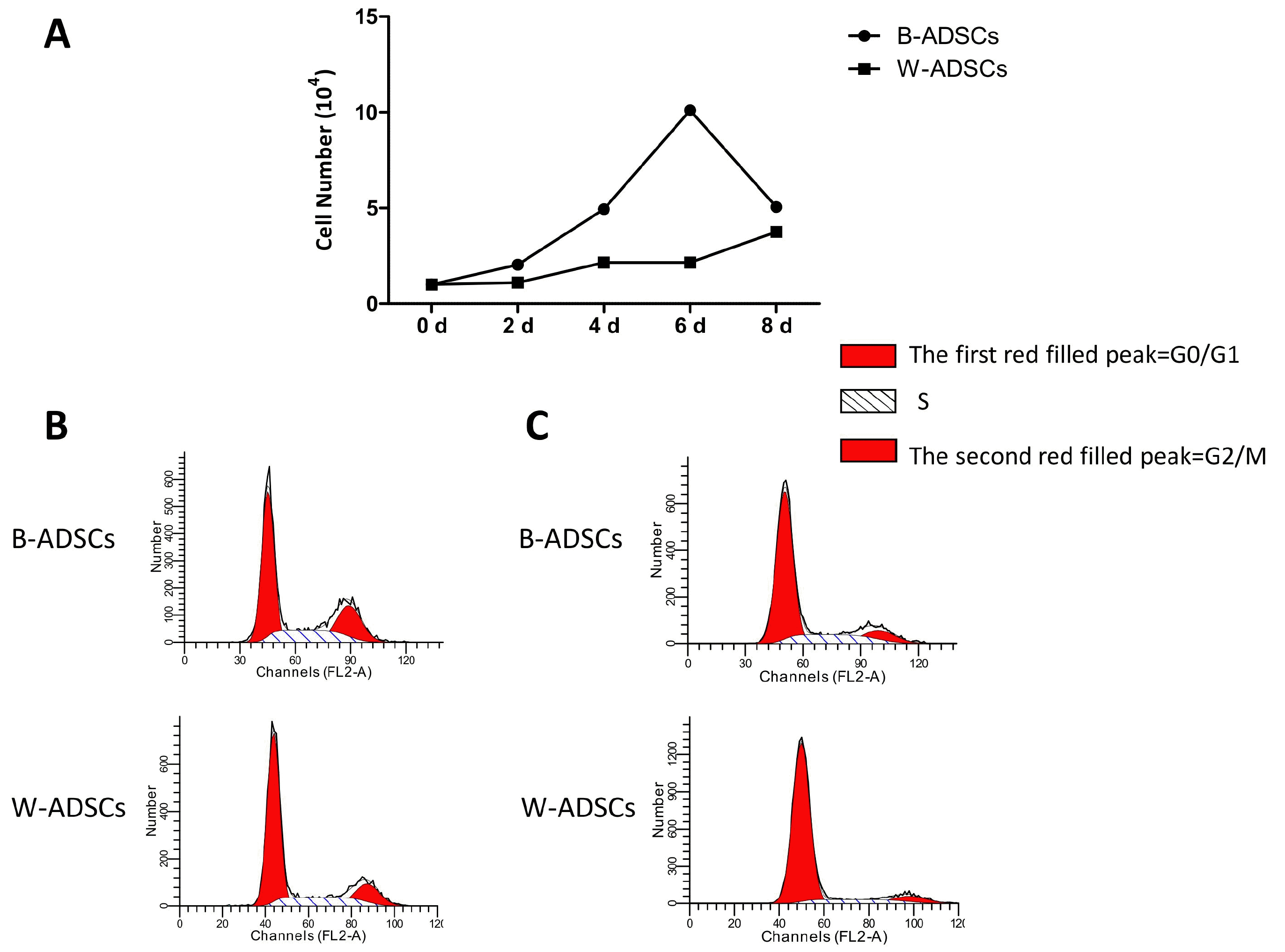
B-ADSCs showed stronger proliferation capacity than W-ADSCs
Next, we examined the proliferation of B-ADSCs and W-ADSCs from young mice (3∼4 weeks) by cell counting assay and cell-cycle phase testing. In the cell counting assay, cells were counted every two days for 8 days. We found proliferation of B-ADSCs was faster than that of W-ADSCs especially at the fourth day (Fig. 2A). In addition, we analyzed the cell-cycle phase via propidium iodide (PI) staining by flow cytometry. We found a higher proportion of B-ADSCs in G2/S phase and a lower proportion in G0/G1 phase than those of W-ADSCs (Fig. 2B). The same result about cell-cycle phase was obtained in ADSCs of aged mice (7∼8 months) (Fig. 2C).
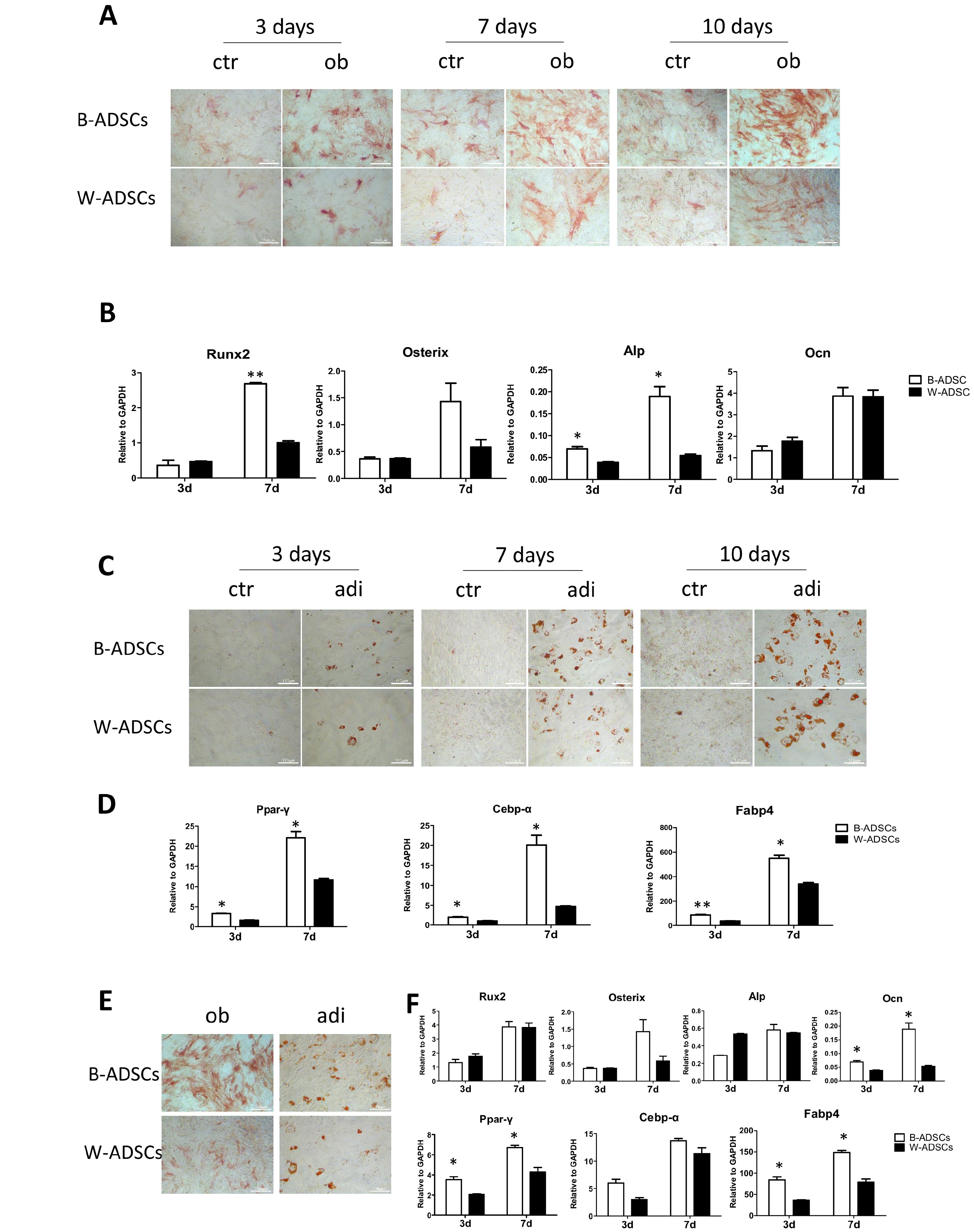
Stronger osteogenic and adipogenic differentiation capacities of B-ADSCs compared with W-ADSCs
As previously reported, ADSCs could differentiate into adipocytes and osteoblasts in vitro when cultured under appropriate conditions (9). Our results showed that B-ADSCs displayed stronger osteogenic and adipogenic differentiation ability. ALP staining and oil drops of B-ADSCs were much more than those of W-ADSCs (Fig. 3A and 3C). These significant augment in osteogenesis and adipogenesis were further observed through the analyses of Runx2, Alp, Ocn, Osterix, Ppar-γ, Cebp-α, and Fabp4 mRNA expression levels (Fig. 3B and 3D). Additionally, in aged mice, osteogenesis and adipogenesis capacities of B-ADSCs were also stronger than those of W-ADSCs via ALnP and Oil-Red-O staining (Fig. 3E) as well as via mRNA expression levels of bone and adipose related genes (Fig. 3F).
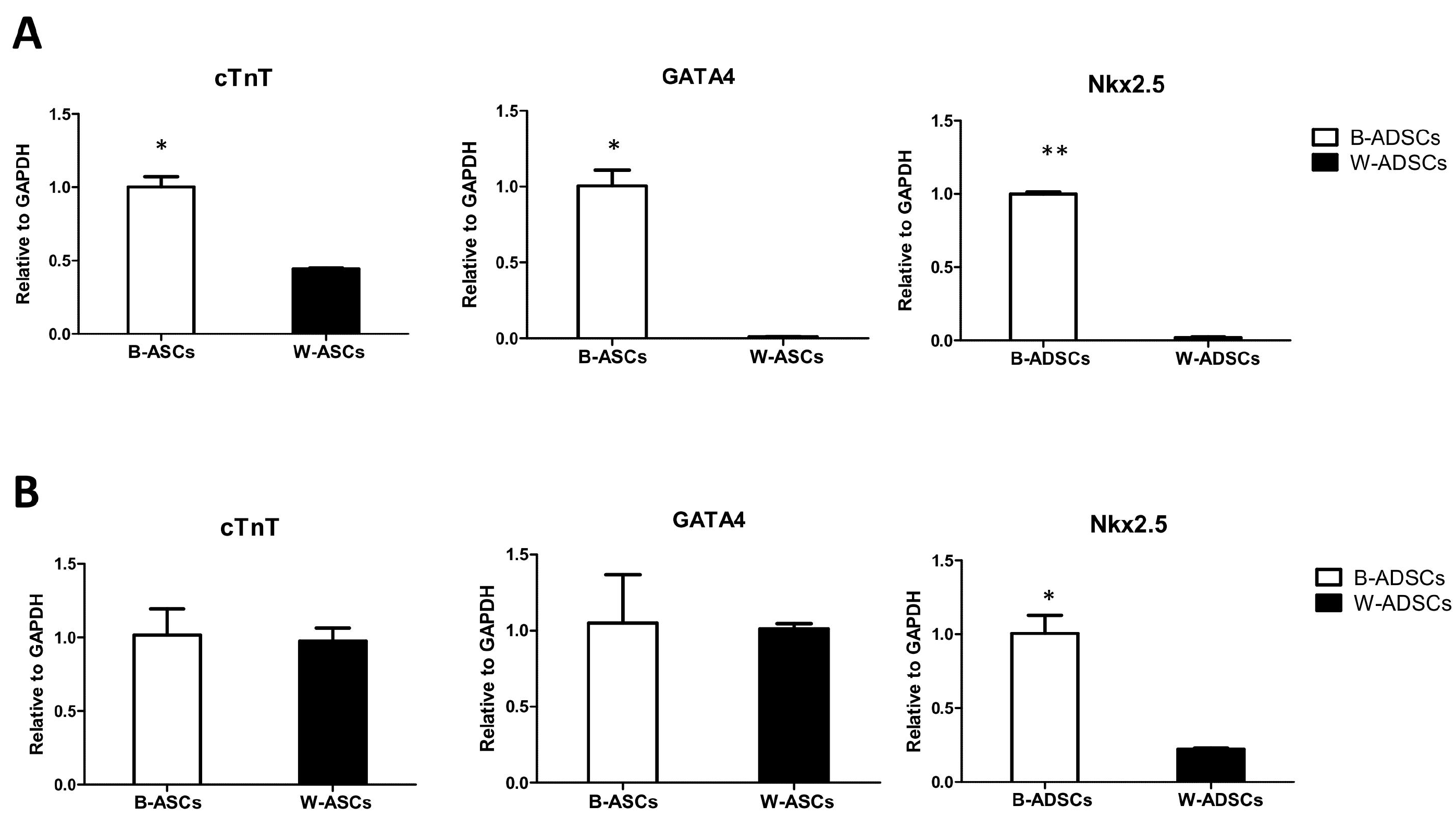
Stronger myocardial differentiation of B-ADSCs compared with W-ADSCs
As is well-known, B-ADSCs could differentiate into cardiomyocyte. So we planted B-ADSCs and W-ADSCs on CNT and examined cardiomyocyte-related gene expression. The results showed that cardiomyocyte-specific markers cTnT and transcription factor GATA4 and Nkx2.5 expressed in B-ADSCs significantly more than those in W-ADSCs (Fig. 4A). This indicated that B-ADSCs did have increased myocardial differentiation capacity as compared to W-ADSCs. In aged mice, only transcription factor Nkx2.5 expressed in B-ADSCs significantly more than in W-ADSCs (Fig. 4B).
Similar immunophenotype of ADSCs
Many molecules such as PD-L1, VCAM-1, ICAM-1 and iNOS have been described to have major roles in MSC-mediated immunosuppression. To investigate the immunophenotype of ADSCs, we treated B-ADSCs and W-ADSCs with IFN-γ and TNF-α at increasing concentrations, and left one group untreated. We found that PD-L1, VCAM-1, and ICAM-1 were significantly up-regulated in ADSCs that were stimulated with IFN-γ and TNF-α at concentration of 2 ng/ml, with no difference observed between B-ADSCs and W-ADSCs (Fig. 5A and 5B). However, PD-L1 was significantly up-regulated in W-ADSCs without stimulation than in B-ADSCs (Fig. 5A). We then compared the mRNA levels of representative inflammatory cytokines (iNOS, TNF-α, IL10, socs1) between B-ADSCs and W-ADSCs by qRT-PCR. without stimulation, both B-ADSCs and W-ADSCs exhibited low levels of iNOS,TNF-α, and socs1 production; while with stimulation, their levels were elevated. However, there was no significant difference between B-ADSCs and W-ADSCs (Fig. 5C). We also compared the immunophenotype of B-ADSCs and W-ADSCs from aged mice, there was no significant difference (data not shown). Taken together, these results indicated that B-ADSCs and W-ADSCs have similar immunoregulation capacity.
Different immunoregulatory capacities of B-ADSCs and W-ADSCs from aged mice
Next, to investigate whether the immunoregulation capacity of B-ADSCs and W-ADSCs in vitro were different, we selected CD3+ T/CD4+ T cells which were labeled with CFSE, and stimulated them with PMA and ionomycin and co-cultured them with B-ADSCs and W-ADSCs, respectively. Changes in CFSE signals were then measured via flow cytometry to monitor T cell proliferation. We found that T cell proliferation was inhibited by both cells, which was indicated by a slower reduction in CFSE fluorescence intensity. Furthermore, a slight augment in T cell proliferation were observed in the one co-culture with W-ADSCs compared to the one co-culture with B-ADSCs at the highest ADSCs-to-T cell ratio. The data indicated that ADSCs reduced the proliferation of CD3+ (Fig. 6A) and CD4+ (Fig. 6B) T cells, but there was no significant difference between B-ADSCs and W-ADSCs in the immunoregulation capacity of young mice. However, B-ADSCs from aged mice showed lower immunosuppression to CD3+ T cells than W-ADSCs at different ratio (ADSCs to T cell) (Fig. 6C).
Discussion
This is the first study to demonstrate that murine B-ADSCs have enhanced proliferation property and osteogenic, adipogenic and myocardial differentiation ability, but have no significant difference in immunoregulation capacity compared to murine W-ADSCs. However, these capacities of B-ADSCs and W-ADSCs change with age.
Adipose tissue is an attractive cell source for stem cell-based treatment because of its self-renewal and multipotential differentiation capacity as well as the ease to harvest in sufficient quantities (22). However, there are several regions around the body where adipose tissue is stored and can be used for isolation of ADCSs. It is confirmed that ADCSs from different regions of adipose tissue show different characteristics (23-25). Few studies have compared the differentiation and immunoregulation capacity of murine ADSCs from BAT and WAT, so the factors that affect these capacities are still unknown. Therefore, it is important and necessary to investigate which region is the best candidate for cell isolation and to understand whether age is an important factor to optimize clinical applications and fundamental researches.
In this study, firstly we isolated B-ADSCs and W-ADSCs from interscapular adipose tissue and inguinal subcutaneous adipose tissue, respectively, and cultured successive passages from passage 0 (P0) to passage 5 (P5). Our results revealed that both B-ADSCs and W-ADSCs showed spindle-shaped, fibroblast-like morphology and similar cell surface marker expression, which conformed to the characteristics of MSCs. Meanwhile, ADSCs from aged mice exhibited the same result. However, the proliferation of B-ADSCs was greater than W-ADSCs both in cell counting and cell-cycle phase assay, suggesting that the proliferating capacity of murine ADSCs is influenced by the harvesting region. Additionally, cell-cycle phase assay of B-ADSCs and W-ADSCs from elder mice also supported the above conclusion. What’s more, a lot of evidence agrees with the role of the harvesting region as a crucial factor that influences cell proliferation rate (24, 26). In our in vitro differentiation experiment, B-ADSCs demonstrated an accelerated osteogenic and adipogenic capability compared with W-ADSCs, confirmed by ALP and Oil Red O staining, respectively. The same results were obtained in ADSCs from 7-to-8-month mice. In addition, B-ADSCs’ significant enhanced osteogenic and adipogenic capability was confirmed by further mRNA expression levels analysis of bone related genes - namely, Alp, Runx2, Ocn, and osterix (27), and adipose related genes - namely, Ppar-γ, Cebp-α, and Fabp4. This difference was also mentioned in a study by Ardeshirylajimi et al. (20), despite different species. Interestingly, we found B-ADSCs from young mice expressed higher levels of earlier phases of osteogenesis (Runx2 and Alp), while B-ADSCs from aged expressed higher levels of late osteogenesis genes, such as Ocn. This difference requires further study. All above indicated that cell-specific differences between B-ADSCs and W-ADSCs did influence the proliferation and osteogenic and adipogenic differentiation of cells.
It has been reported that adipose tissue contains stem cells that have the ability to differentiate into cardiomyocyte (28, 29). Therefore, we analyzed cardiac differentiation markers GATA4, Nkx2.5 (30) and cTnT mRNA expression of B-ADSCs and W-ADSCs. We found that the expression of cTnT and transcription factor GATA4 and Nkx2.5 in B-ADSCs were significantly more than those in W-ADSCs. However, only transcription factor Nkx2.5 expressed in B-ADSCs from aged mice was significantly more than in W-ADSCs. It suggested that B-ADSCs from young mice may be an appropriate candidate of the cell source for cardiomyocyte regeneration. Nonetheless, it requires more studies to understand whether myocardial differentiation of B-ADSCs attenuate with age.
To our knowledge, no report identifies the immunomodulation capacity between murine B-ADSCs and W-ADSCs. So we sought to determine if different results could be seen in the two cells from different regions. Several molecules such as PD-L1, ICAM-1, VCAM-1, iNOS and IL10 have been proposed in the mechanisms of immunosuppression by mouse MSCs (31-33). Our results showed that PD-L1 was up-regulated more significantly in W-ADSCs without stimulation than that in B-ADSCs, but this difference disappeared with the increase of stimulation concentration. The expression of ICAM-1 and VCAM-1 had no differences between B-ADSCs and W-ADSCs. It is important to note, B-ADSCs maybe show potential immunosuppression capacity and are needed further deep study. However, the expression of iNOS, IL10 and TNF-α showed no significant difference. Similarly, there was no difference between B-ADSCs and W-ADSCs in socs1, which controls signaling of many cytokines, including TNF-α and IL-10 with a feedback loop mechanism (34). In addition, in CFSE assay, both B-ADSCs and W-ADSCs inhibited the proliferation of CD3+ and CD4+ T cells as reported in many reports (35-37). Immunosuppression capacity to T cells between B-ADSCs and W-ADSCs had no significant difference. However, B-ADSCs from aged mice showed lower immunosuppression capacity to CD3+ T cells than W-ADSCs from aged mice.
Overall, this study identified the difference between murine B-ADSCs and W-ADSCs about their proliferation, differentiation and immunosuppression capacity. More importantly, these capacities would change with age. Hence, it provides new insights into the influence of ADSCs derived from different regions and ages, which would provide evidence for using tissue-specific ADSCs that are applicable for cell therapy and tissue engineering in murine species. Nonetheless, the deep molecular mechanisms of the difference still require further studied.
 XML Download
XML Download