

 PDF
PDF Citation
Citation Print
Print


Introduction
Due to the widespread use of mechanical ventilation and hyperoxia, the fetal survival rate has greatly improved; however, the incidence of bronchopulmonary dysplasia (BPD) has also increased. BPD majorly affects the growth, development, and quality of life of premature infants. Although hormone treatments can reduce inflammation, the use of hormones also increases the risk of hypertension, hyperglycemia, gastrointestinal bleeding, intestinal perforation, and hypertrophic obstructive cardiomyopathy in newborns (1). Hormones also show direct toxicity towards the developing brain of newborns, which can lead to neuron necrosis. Hormones can also interfere with and inhibit brain development (2). Exogenous pulmonary surfactant (PS) can promote alveolar development, improve lung function, and reduce the severity of BPD. However, PS is only effective over a short-term period, and is not effective for the long-term improvement of lung function (3). In addition, treatment with vitamin A (4), superoxide dismutase (SOD), erythropoietin, nitric oxide inhalation, and caffeine (5) have also showed protective anti-inflammatory effects on brain development; however, there is no conclusive evidence confirming that these methods have long-term beneficial effects on lung development. Therefore, new effective therapies for BPD are urgently required.
In earlier papers reported by our laboratory, we found that umbilical cord mesenchymal stem cells (UC-MSCs) can improve the survival rate of rats with acute lung injury induced by lipopolysaccharide (LPS), and reduced inflammatory cell infiltration and inflammatory factors in the lungs (6). Thus, UC-MSCs may show a therapeutic basis in promoting anti-inflammatory homeostasis and reducing oxidative stress. Recently, many animal models and clinical trials have also shown that MSCs can reduce pulmonary fibrosis, improve lung structure, and improve the survival rate in BPD patients. MSCs play an important role in coordinating the growth of epithelial cells and endothelial cells in lung development (7, 8). Moreover, recent phase I clinical trials and 2-year follow-up have shown that allogeneic bone marrow mesenchymal stem cell transplantation for BPD in premature infants is feasible, and its short-term and long-term safety has also been demonstrated (8, 9). However, the mechanism of MSCs in the treatment of BPD is still unclear, and its efficiency needs to be further improved. It is thus important to identify stem cells that are more effective for BPD treatment and to explore the molecular basis of their mechanism of action.
Human amnion-derived mesenchymal stem cells (hAD-MSCs) not only have the characteristics of MSCs, but also express several characteristics similar to embryonic stem cells (10). In addition, the method for obtaining hAD-MSCs is safe, non-invasive and ethical for both mothers and infants (11). In this study, we evaluated whether the intratracheal transplantation of hAD-MSC-derived exosomes (EXOs) would be as effective as treatment with hAD-MSCs alone in a neonatal hyperoxic lung injury (NHLI) rat model. We further identified differentially expressed genes in lung tissues before and after hAD-MSC treatment.
Go to : 
Materials and Methods
Preparation of hAD-MSCs
Human placentas (n=10; normal clinically term pregnancies) were collected from healthy donors who received cesarean sections at the Qilu Hospital of Shandong University, China. The use of placentas was approved by the Ethics Committee of Qilu Hospital of Shandong University, and written informed consent was obtained from all donors.
Under sterile conditions, the amniotic membrane was stripped from the placenta and cut into fragments with sharp scissors. At 37℃, the debris is digested with 0.05% trypsin (Sigma Aldrich, St. Louis, MO, USA) for 2 h with shaking. The tissue debris obtained after digestion was then centrifuged at 120 ×g for 10 min. The supernatant was discarded and amniotic debris was re-digested in 2 mg/mL type II collagenase (Sigma Aldrich) containing 0.1 mg/mL DNase I (Sigma Aldrich) and 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at 37℃ with shaking. The undigested tissue was removed by filtering. The filtrate was washed with Hanks’ Balanced Salt Solution (HBSS) and resuspended in complete media composed of Dulbecco’s modified Eagle’s medium (DMEM)/F-12 supplemented with 10% FBS, 100 U/mL penicillin, and 100 g/mL streptomycin (Gibco) in 100% humidity with 5% CO2 and 21% O2 at 37℃. hAD-MSCs at the passage 3 were used for subsequent tests.
Cell phenotyping of hAD-MSCs
Flow cytometry was performed to characterize hAD-MSCs. The following phycolerythrin (PE)-labeled antibodies were used to analyze cell phenotypes: anti-human CD29 (557332), CD31 (560983), CD34 (550761), CD44 (562818), CD45 (560975), CD73 (562430), CD90 (561970), and CD105 (560839) (all diluted 1:10; BD Co., San Diego, CA, USA). Next, 1×106 cells were stained with 10 μL each antibody in 100 μL phosphate-buffered saline (PBS) for 15 min at room temperature. Flow cytometry was performed using the Guava easyCyte 6HT (EMD Millipore, Billerica, MA, USA), and the data were examined with Guava Incyte software (version 2.8, EMD Millipore).
Differentiation capacity
To investigate the differentiation ability of these adherent cells, passage 4 cells were cultured under corresponding conditions according to previous published literature (12). Cells were cultured at a density of 2×104 cells/cm2 and replaced with induction medium. Osteogenic differentiation medium consisted of L-DMEM supplemented with 10% FBS, 0.1 μM dexamethasone, 50 mM β-glycerol phosphate, and 0.2 mM ascorbic acid (Sigma Aldrich). The basic medium for adipose-induced differentiation is high-glucose DMEM. Additional compounds include 0.25 mM 3-isobutyl-1-methylxanthine, 0.1 μM dexamethasone, 0.1 mM indomethacin (Sigma Aldrich), 6.25 μg/mL insulin (PeproTech, Rocky Hill, NJ, USA), and 10% FBS (Gibco). Cells grown in normal growth medium served as the control. To induce chondrogenic differentiation, cells were transferred into 15-mL polypropylene tubes and centrifuged at 240 ×g for 5 min; the pellet was collected and treated with chondrogenic medium for 3 weeks. Chondrogenic medium consists of high-glucose DMEM (Bio-Fluid Technologies, Rockville, MD, USA) supplemented with 0.1 μM dexamethasone, 50 μg/mL ascorbic acid, 100 μg/mL sodium pyruvate, 40 μg/mL proline (Sigma Aldrich), 10 ng/mL transforming growth factor (TGF)-β1, and 50 mg/mL ITS+ premix (6.25 μg/mL insulin, 6.25 μg/mL transferrin, 6.25 ng/mL selenius acid, 1.25 mg/mL bovine serum albumin (BSA), and 5.35 mg/mL linoleic acid; BD Co.). The differentiation media were changed every 3-4 d.
To detect adipose orientation induced differentiation, cells were fixed and stained with oil-red O (Sigma Aldrich) for 10 minutes. Chondrogenic differentiation was evaluated with anti-Collagen II antibody (ab34712, dilution 1:250, Abcam, Cambridge, UK) for 45 min at 37℃, and then peroxidase-conjugated secondary antibodies (CWBIO, dilution 1:200) were added for 30 min at room temperature. The cells were then incubation with DAB chromogen (CWBIO) and counterstained with haematoxylin. For evaluation of mineralized matrix, cells were stained with Alizarin-red S (Sigma Aldrich). In addition, osteogenic differentiation was also stained with chromogenic alkaline phosphatase activity (ALP) assay kit (Jiancheng Corp, Nanjing, China).
Preparation of conditioned medium
For conditioned medium (CM) collection, fresh full medium was replaced when hAD-MSCs reached 70∼80% confluency. After 24 h, the medium was collected and centrifuged at 1000 ×g for 20 min with a 0.1-μm pore filter to remove detached MSCs and cellular debris. MSC-derived CM was stored at −80℃ until use.
Isolation and identification of EXOs
After hAD-MSCs reached 90% confluency, the cells were washed and serum-starved for 6 h in basal medium (DMEM/F-12; Gibco). Thereafter, the medium was collected and centrifuged at 720 ×g for 30 min at 4℃ to remove cellular debris, followed by centrifugation at 100,000 ×g for 100 min at 4℃ (Sorvall LYNX-6000, Thermo Fisher Scientific) to sediment the EXOs. The protein content of the concentrated EXOs was measured using the bicinchoninic acid (BCA) protein assay kit (CW0014; CWBiotech, Beijing, China). The morphology of the exosomes was observed using transmission electron microscopy (TEM; JEOL-1200-EX II; JEOL, Tokyo, Japan). Exosomes were identified using specific antibodies targeting CD63 (ab217345, Abcam), CD9 (ab92726, Abcam), CD81 (ab109201, Abcam), and heat shock protein 70 (Hsp70; ab79852, Abcam), which were used for western blotting analysis. All antibodies were diluted to 1:1,000.
Animal model and hAD-MSC therapy
Animal procedures were approved by the Animal Care Unit and Use Committee of Qilu Hospital, China (QL-TXB-2019044). Timed pregnant Sprague-Dawley rats (Charles River Co., Beijing, China) spontaneously delivered rat pups. Newborn rats were allocated to five experimental groups: normoxia control, hyperoxia model, hyperoxia with hAD-MSCs, hyperoxia with hAD-MSC-CM, and hyperoxia with hAD-MSC-EXOs (n=12 per group). Normoxic rat pups were maintained in unmodified air, and hyperoxic rat pups were raised in hyperoxic chambers (80% O2) from birth until postnatal day (P) 14.
At P7, the pups were anesthetized with ketamine and xylazine via intraperitoneal injection. The trachea was exposed through a small midline incision on the neck. hAD-MSCs (1×106 cells/50 μL), hAD-MSC-CM (50 μL), or hAD-MSC-EXOs (300 ng/50 μL) were delivered by tracheal puncture with a 30-gauge needle. Pups were placed in a warmed plastic chamber under normoxic or hyperoxic conditions for recovery. Once the pups were fully awake, they were returned to their dams. The schematic diagram of the experimental process is shown in Fig. 1A. Survival and body weight of rat pups in each group were checked daily throughout the experiment.
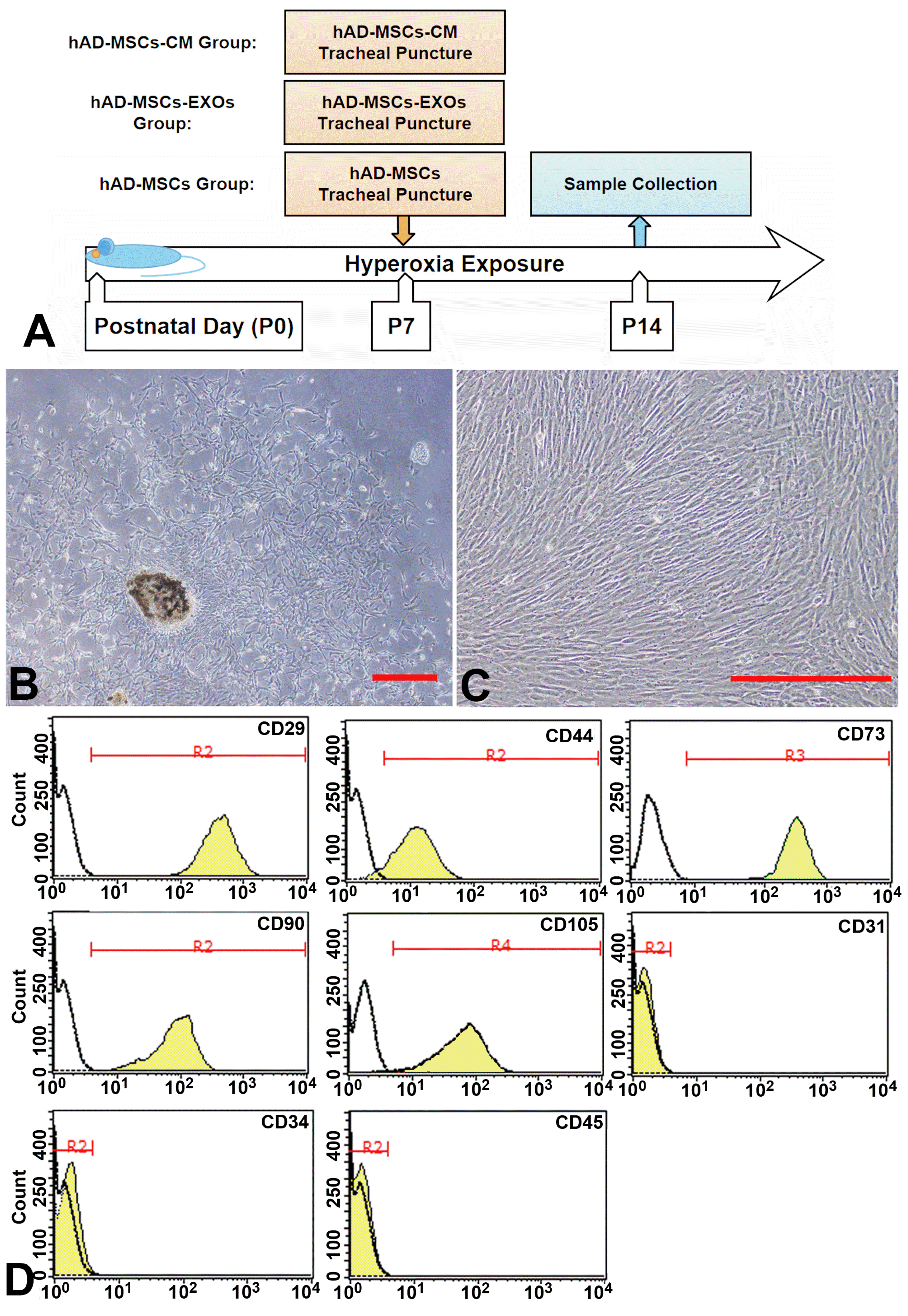 | Fig. 1Characterization of human amniotic mesenchymal stem cells (hAD-MSCs). (A) Overall experimental design process and grouping. (B) Within days of primary culture, small debris of amniotic membrane usually become germinal centers of adherent cells. (C) Adherent cells growing from amniotic tissue mass showed typical fibrous morphology and grew tightly. Bar=100 μm. (D) Flow cytometry showed that the cells positively expressed MSC markers (CD29, CD44, CD73, CD90, and CD105), but negatively expressed CD31, CD34, and CD45.
|
Collection of bronchoalveolar lavage fluid and tissue samples
After euthanizing the rats pups on P14, the right bronchial tube was ligated. A 20-gauge catheter and 1 mL cold PBS were used to flushed back and forth three times from trachea to got bronchoalveolar lavage fluid (BALF). The BALF fluid was centrifuged at 720 ×g for 15 min at 4℃. The resulting pellet was used to make cell smears and Wright-Giemsa staining was used to count neutrophils. Protein concentration in BALF was then detected by Bio-Rad BCA protein Quantitative Assay Kit (Bio-Rad Laboratories, Hercules, CA, USA). The right middle lobe was preserved in liquid nitrogen at −80℃ for subsequent gene expression analysis. The right upper lobe is used to compare the degree of pulmonary edema. The right lower lobes were used for pathological evaluation.
Cytokine measurement in BALF
Enzyme-linked immunosorbent assay (ELISA) was used to measure the concentration of tumor necrosis factor-α (TNF-α, R&D Systems, Minneapolis, MN, USA), Interleukin-1 beta (IL-1β, Boyun, Shanghai, China), IL-6 (Boyun), and monocyte chemoattractant protein-1 (MCP-1, Boyun) in BALF according to the manufacturer’s instructions. The ELISA plate was read at 450 nm and analyzed according to the standards.
Malondialdehyde concentration in BALF
Malondialdehyde (MDA) levels in the BALF, the indicator of lipid peroxidation, were detected using a thiobarbituric acid (TBA) photometric Kit (A003-1, Jchio, Nanjing, China) according to the manufacturer’s procedures. The absorbance of the organic layer was measured via spectrophotometry at 532 nm (Pharmacia Biotech, Fleming Island, FL, USA). MDA levels were expressed in nmol/mL.
Lung histopathology
For histological analysis, formalin fixed and paraffin-embedded lung tissue was cut into 5-μm sections and stained with hematoxylin and eosin. The injury status of each lung were blindly scored by a pathologist in terms of the following four categories: alveolar congestion, hemorrhage, neutrophil infiltration into the airspace or vessel wall, and thickness of alveolar wall/hyaline membrane formation. Each sample was graded on a 0- to 4-point scale: 0=no injury; 1=injury in up to 25% of the field; 2=injury in up to 50% of the field; 3=injury in up to 75% of the field; and 4=diffuse injury (13). To perform standardized analysis through lung morphometry, the mean linear intercept (MLI), an indicator of mean alveolar diameter, was assessed in 10 nonoverlapping fields (14).
Lung weight/body weight ratio analysis
The lung tissue was weighed, and then the wet lung weight was divided by body weight to get the lung weight/body weight ratio.
Superoxide dismutase activity assay
SOD activity was analysed by spectrophotometry based on nitroblue tetrazolium (NBT) method (S0109, Beyotime, Jiangsu, China) according to the manufacturer’s procedures. The amount of formazan formed was calculated in absorbance measurements at 560 nm (Pharmacia Biotech) and was expressed in U/mL.
Microarray-based gene expression analysis
TRIzol (Ambion, Life Technologies, Carlsbad, CA, USA) were used to extract total RNA from lung tissue according to the manufacturer’s procedures. RNA labeling and hybridization were performed according to the Agilent One-Color Microarray-Based Gene Ex pression Analysis protocol (Agilent Technologies, Santa Clara, CA, USA). Acquired array images were analysed using Agilent Feature Extraction software (version 11.0.1.1). Quantile normalization and subsequent data processing were performed using the GeneSpring GX v12.1 software package (Agilent Technologies). Differentially expressed genes between the two samples were identified through Fold Change filtering. Through the Kyoto Encyclopedia of Genes and Genomes (KEGG; www.genome.jp/kegg/pathway.html) pathway enrichment analysis, significantly enriched pathways were identified in differentially expressed genes (DEGs) with a fold change ≥two.
Quantitative PCR validation of microarray expression data
Total RNA was extracted from each sample using TRIzol (Invitrogen, Carlsbad, CA, USA). Reverse-transcriptase quantitative PCR (qRT-PCR) was performed to analyze the expression levels of ten genes selected from the above microarray results (the primers used are listed in Table 1). Total RNA was reverse-transcribed to cDNA using the Omniscript cDNA Synthesis Kit (Qiagen, Hamburg, Germany) according to the manufacturer’s instructions. qRT-PCR was conducted using an PCR system (Analytik Jena AG, qTOWER3G, Germany) and SYBR green I dye (Toyobo, Osaka, Japan). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal reference control. The expression of each gene was determined using the 2−DDCT method. The qPCR conditions were as follows: 40 cycles of 95℃ for 4 min, 94℃ for 15 s, and 60℃ for 1 min. Data are reported as the mean±standard deviation (SD) of at least three independent experiments. mRNA expression is presented as fold difference with respect to normoxia groups.
Table 1
Primers used in qRT-PCR
![]()
Western blot analysis
Western blots were carried out to analyze the expression of acylaminoacyl-peptide hydrolase (APEH). After electrophoresis and transfer, the membranes were incubated with the primary antibodies anti-APEH (ab236608, dilution 1:2000, Abcam) and anti-actin (ab8227, dilution 1:1000, Abcam), followed by incubation with HRP-conjugated secondary detected by enhanced chemiluminescence (ECL; Beyotime) and quantified using C-Digit (Model: 3600, Image Studio Digits Ver 4.0; Licor, Lincoln, NE, USA).
Statistical analysis
Data were analyzed using SPSS software version 17.0 (SPSS Inc., Chicago, IL, USA). Quantitative data are described as the mean±SD. Data were compared by analysis of variance (ANOVA) or Student’s t-test. A value of p< 0.05 was considered statistically significant.
Go to :
Results
Characterization of hAD-MSCs and their differentiation capacity
Initially, amniotic membranes were digested using trypsin for 2 h; the remaining tissue was digested by collagenase II. The digested fragments quickly grew into fibroblasts attached to the wall of cell culture flask (Fig. 1B). These adherent cells showed a typical fibrous morphology and grew rapidly, with an average doubling time of 36.4 h at passage 5 (Fig. 1C). Flow cytometry showed that these cells positive express MSCs markers (CD29, CD44, CD73, CD90, and CD105); and negative express endothelial cell markers (CD31) and hematopoietic markers (CD34 and CD45) (Fig. 1D).
In adipogenic induction group, almost all cells become hypertrophic after 2 weeks. A large number of lipid droplets can be dyed red by oil red O in the cytoplasm (Fig. 2A). Immunohistochemical staining of type II collagen, marker for chondrocytes, also showed positive results after 21 days of chondrogenic induction (Fig. 2B). After 2 weeks of osteogenic induction, cell aggregation increases and can be stained positive by Alizarin red, which means the formation of mineralized matrix (Fig. 2C), and most MSC-like cells were alkaline phosphatase-positive by the end of 14 d (Fig. 2D).
 | Fig. 2Differentiation capacity of hAD-MSCs and characterization of hAD-MSC-derived exosomes (EXOs). (A) After adipogenic induction, almost all cells became broad and contained fat droplets dyed by oil red O. (B) After 21 days chondrogenic induction, hAD-MSCs expressed high concentrations of type II collagen, with a brown signal. (C) Under osteogenic conditions, MSCs aggregated, and were stained with Alizarin red, and (D) most cells were found to be alkaline phosphatase-positive, with a red signal. (E) The average diameter of the exosomes was 110 nm. (F) Western blotting indicated that hAD-MSC-EXOs expressed exosomal markers such as CD63, CD9, Hsp70 and CD81. Magnification: 100×.
|
Characterization of hAD-MSC-EXOs
Serum starvation was used to promote the secretion of exosomes, and exosomes were then precipitated by high-speed centrifugation. The protein concentration was measured to quantify exosomes; an average of 150 ng exosomes per 100 mL medium was obtained. TEM demonstrated that the hAD-MSC-EXOs had diameters ranging from 30∼150 nm (Fig. 2E). Additionally, we confirmed the expression of the extracellular vesicle (EXOs) markers CD9, CD63, CD81, and Hsp-70 by western blotting (Fig. 2F).
hAD-MSCs more effectively alleviated the inflammatory response in NHLI rats
Exposure to oxygen (HC) significantly reduced the survival rate to 41.6% (p<0.05 vs. Normoxia group) at the end of experiment (P14) compared to the 100% survival rate of Normoxia group. After cell infusion on day P7, the survival rate of the hAD-MSCs group was significantly improved (p<0.05 vs. Hyperoxia group), and no further death occurred. However, survival rates of hAD-MSC-EXOs group and hAD-MSC-CM group were lower than that of hAD-MSCs group (Fig. 3A). Hyperoxia exposure also significantly reduced the vitality of the experimental animals; they showed signs of depression, and displayed reduced activity and dietary intake. Additionally, their fur became dull and they showed reduced body weight. Neither cell therapy nor cell-derivative therapy could significantly increase the weight gain of neonatal rats in the experimental group (Fig. 3B). However, the direct transfusion of cells and the transfusion of cell derivatives all successfully reduced the ratio of lung weight/body weight (Fig. 3C) and reduced pulmonary edema, which was further confirmed in tissue sections (Fig. 4A∼E). We used a five-level quantitative scoring system and MLI value to evaluate the therapeutic effect of hAD-MSCs on NHLI. Hyperoxia resulted in the inflammatory cell infiltration, alveolar septum thickened and the structural destruction of the lung parenchyma. As shown in Fig. 4F and 4G, the hyperoxia group exhibited significantly higher lung injury scores and MLI values than the normoxia group. hAD-MSCs, hAD-MSC-EXOs, and hAD-MSC-CM significantly alleviated pulmonary edema, especially in the hAD-MSCs group (p<0.05 vs. Hyperoxia group).
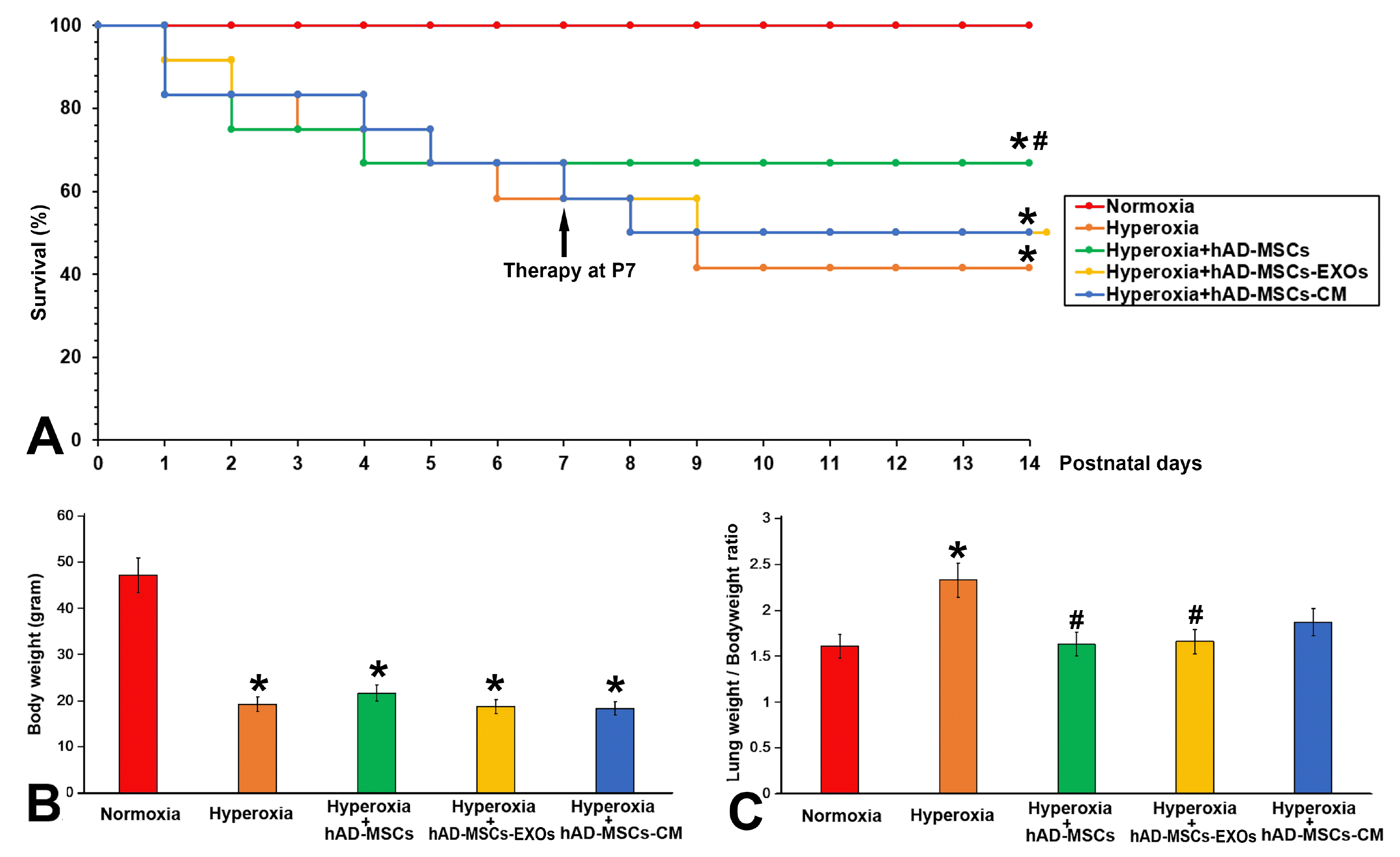 | Fig. 3hAD-MSCs effectively improved survival and body weight. (A) Survival curve. (B) Hyperoxia exposure significantly reduced the body weight of neonatal rats. Treatment with hAD-MSCs and their derivatives did not significantly increase the body weight of neonatal rats. (C) Hyperoxia exposure significantly increased the lung weight/body weight ratio. Both the direct transfusion of cells and the transfusion of cell secretions reversed this increase. At P14: *p<0.05 vs. Normoxia group, #p<0.05 vs. Hyperoxia group.
|
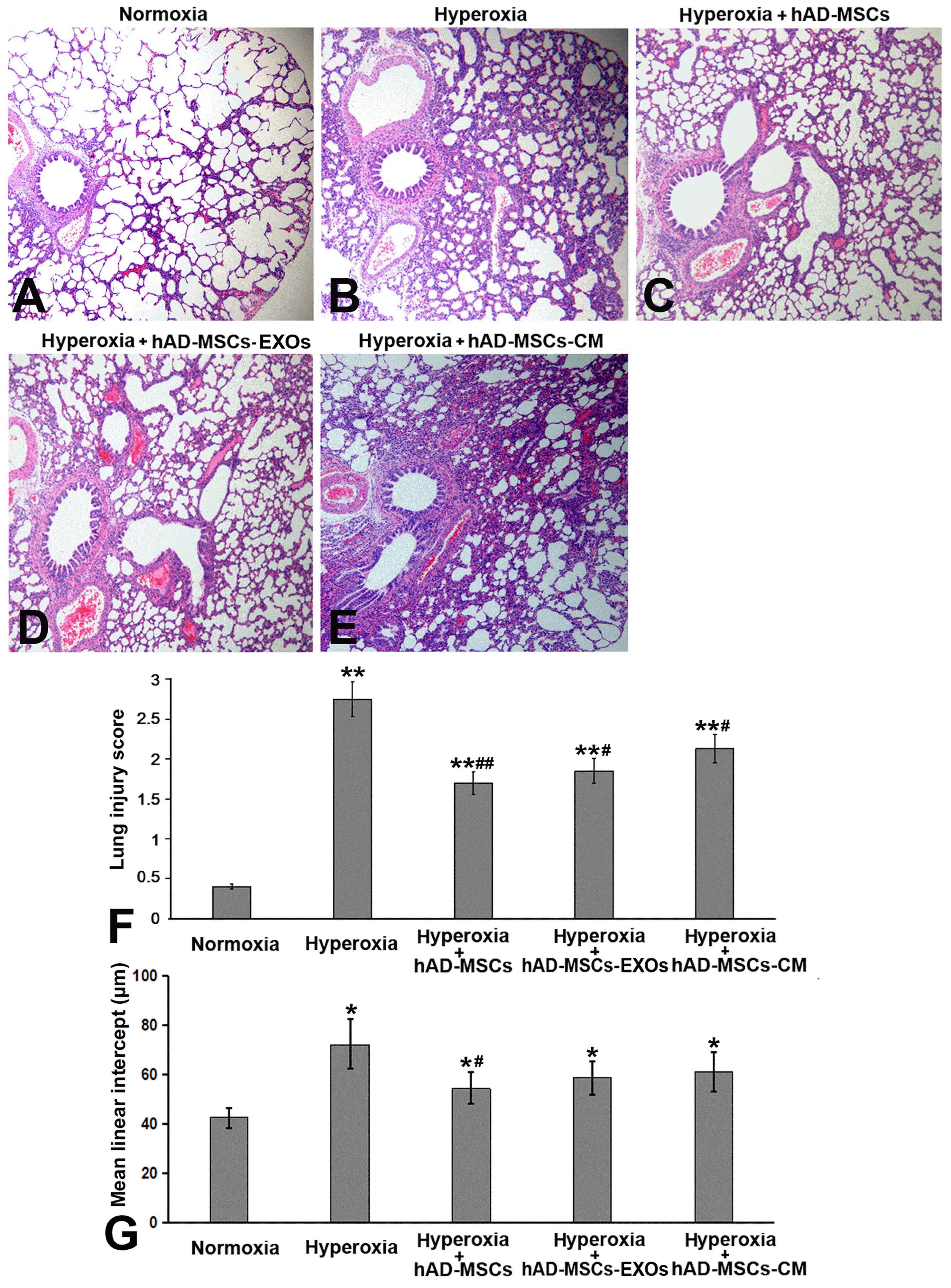 | Fig. 4Histological analysis of neonatal hyperoxic lung injury (NHLI) lungs. (A∼E) Compared to normoxia exposure (A), hyperoxia exposure (B) caused capillary expansion and alveolar hyperemia, as well as infiltration of neutrophils. The direct transfusion of cells (C), and the transfusion of exosomes (EXOs) (D) and conditioned media (CM) (E) reduced pulmonary edema. (F) To evaluate the effect of hAD-MSCs on NHLI, we used a 5-level evaluation system. hAD-MSCs, hAD-MSC-EXOs, and hAD-MSC-CM significantly alleviated pulmonary edema, especially in the hAD-MSC-treated group. (G) Mean linear intercept (MLI) values in the five groups. The lung tissue sections were assessed through lung morphometry. The hyperoxia group exhibited significantly higher MLI values than the normoxia and three therapy groups. hAD-MSCs treatment significantly reduced the hyperoxia-induced increase in lung injury scores and MLI values. Magnification: 100×. *p<0.05 vs. Normoxia group, **p<0.01 vs. Normoxia group, #p<0.05 vs. Hyperoxia group, ##p<0.01 vs. Hyperoxia group.
|
We next detected changes of four cytokines (TNF-α, IL-1β, IL-6, and MCP-1) in BALF, and found that hyperoxia exposure led to a significant increase in these four inflammatory mediator; their expression showed the greatest decrease after the infusion of hAD-MSCs. After the infusion of hAD-MSC-EXOs or hAD-MSC-CM, the expression of inflammatory cytokines also showed a significant decrease, but this effect was not as robust as that of the direct tracheal infusion of hAD-MSCs (Fig. 5A∼D). The same trend was reflected in terms of the number of neutrophils, MDA concentration, and total proteins in BALF (Fig. 5E∼G). SOD activity in BALF was increased significantly after treatment with hAD-MSCs (Fig. 5H).
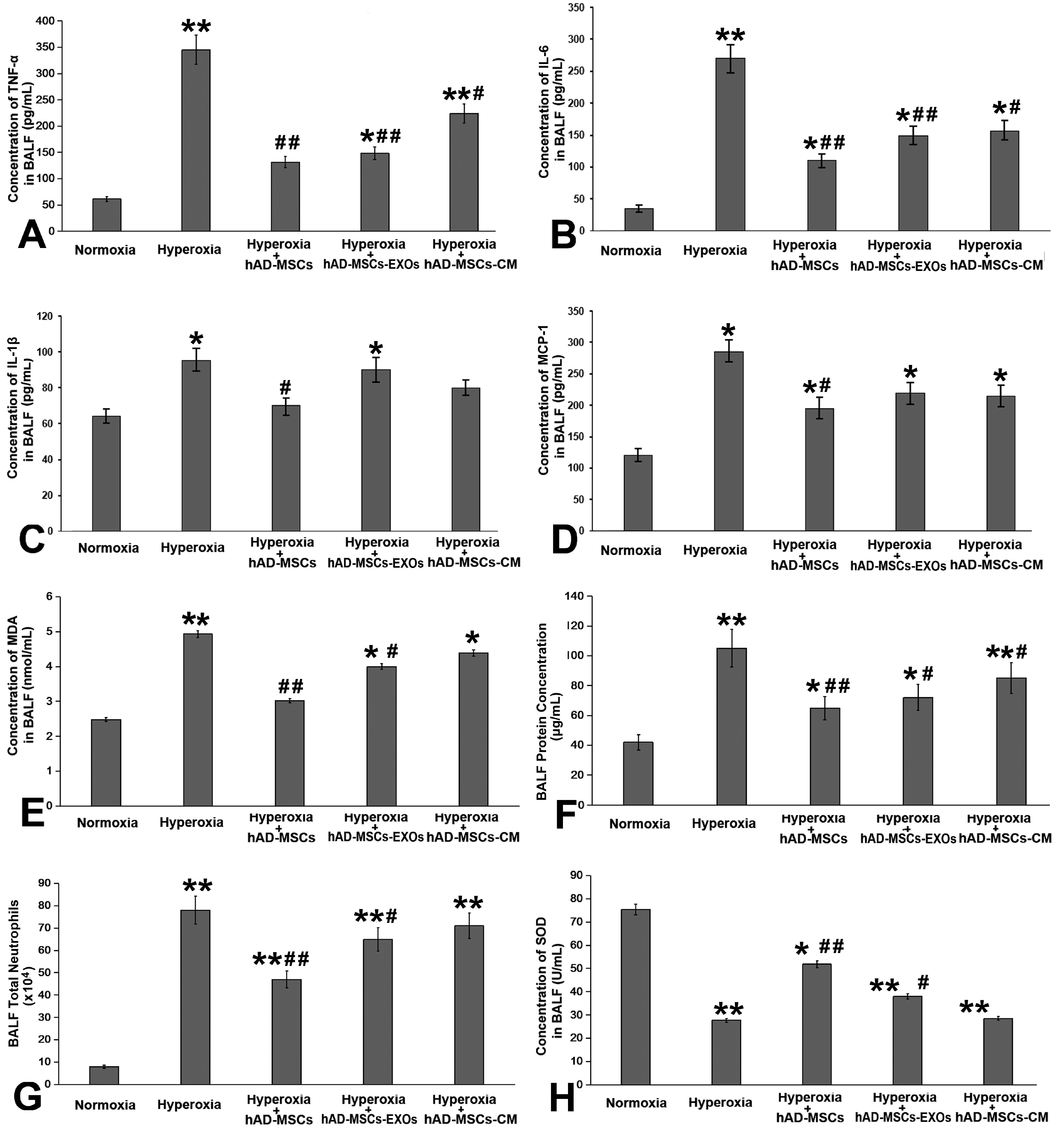 | Fig. 5hAD-MSCs effectively alleviated inflammatory response. Both hAD-MSCs and their derivatives reduced the levels of (A) TNF-α, (B) IL-6, (C) IL-1β, (D) MCP-1, (E) malondialdehyde (MDA), (F) overall proteins, and (G) neutrophils in bronchoalveolar lavage fluid (BALF); these were all increased after hyperoxia exposure. (H) Hyperoxia exposure significantly decreased the expression of superoxide dismutase (SOD) in BALF. Both hAD-MSCs and hAD-MSC-EXOs significantly increased SOD expression, while hAD-MSC-derived conditioned media (CM) did not. *p<0.05 vs. Normoxia group, **p<0.01 vs. Normoxia group, #p<0.05 vs. Hyperoxia group, ##p<0.01 vs. Hyperoxia group.
|
Microarray-based gene expression analysis
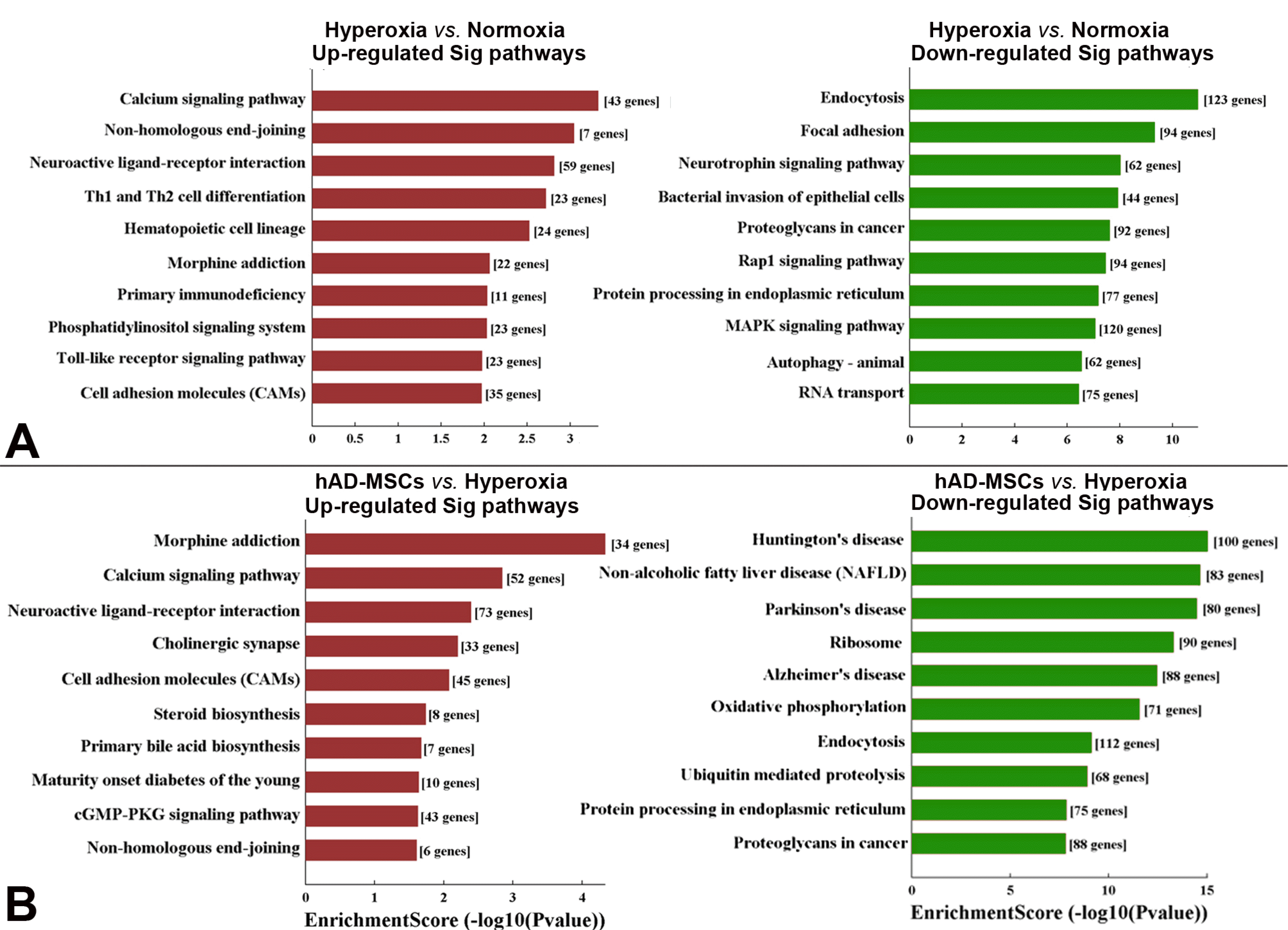
Signaling pathways: We compared differentially expressed genes among the different groups using the obtained lung samples. Compared to the normoxia group, the top three up-regulated signaling pathways in the hyperoxia group were calcium signaling, non-homologous end-joining, and neuroactive ligand-receptor interaction, while the top three down-regulated signaling pathways in the hyperoxia group were endocytosis, focal adhesion, and neurotrophin signaling (Fig. 6A). When treated with hAD-MSCs, the top three up-regulated pathways were morphine addiction, calcium signaling, and neuroactive ligand-receptor interaction compared to those in the hyperoxia group. The top three down-regulated signaling pathways in the hAD-MSCs group compared to those in the hyperoxia group were Huntington’s disease, non-alcoholic fatty liver disease, and Parkinson’s disease (Fig. 6B).
Differentially expressed genes: The expression of four genes was significantly decreased in lungs after hyperoxia injury, but increased significantly after hAD-MSCs treatment: SH3 and multiple ankyrin repeat domains 1 (Shank1), APEH, solute carrier family 26 member 1 (Slc26a1), and protein tyrosine phosphatase 4A3 (Ptp4a3). In addition, the expression of six genes was highly up-regulated after hyperoxia injury, but significantly decreased after hAD-MSCs treatment: ring finger protein 19B (Rnf19b), leucine rich repeat containing 2 (Lrrc2), protein phosphatase, Mg2+/Mn2+ dependent 1B (Ppm1b), family with sequence similarity 96 member A (Fam96a), lactoperoxidase (Lpo), and surfactant protein C (Sftpc). These genes all showed a change in expression of more than threefold.
Changes in APEH expression were closely related to lung injury and hAD-MSC treatment
RT-qPCR analysis was performed to confirm the expression profiles obtained by gene chip analysis. As shown in Fig. 6, the changes in the expression levels of APEH were the most significant (p<0.01, Fig. 7A), and the observed changes were consistent with the results of gene chip analysis. The nine other tested genes were eliminated because either their changes were not significant, or their trends were not consistent with the results of gene expression profiling microarrays. Thus, we selected APEH to further verify changes in its expression at the protein level using western blotting. Through gray value quantification, the data showed that APEH expression decreased significantly after hyperoxia injury (p<0.05), but increased significantly after hAD-MSC treatment (p<0.01, Fig. 7B).
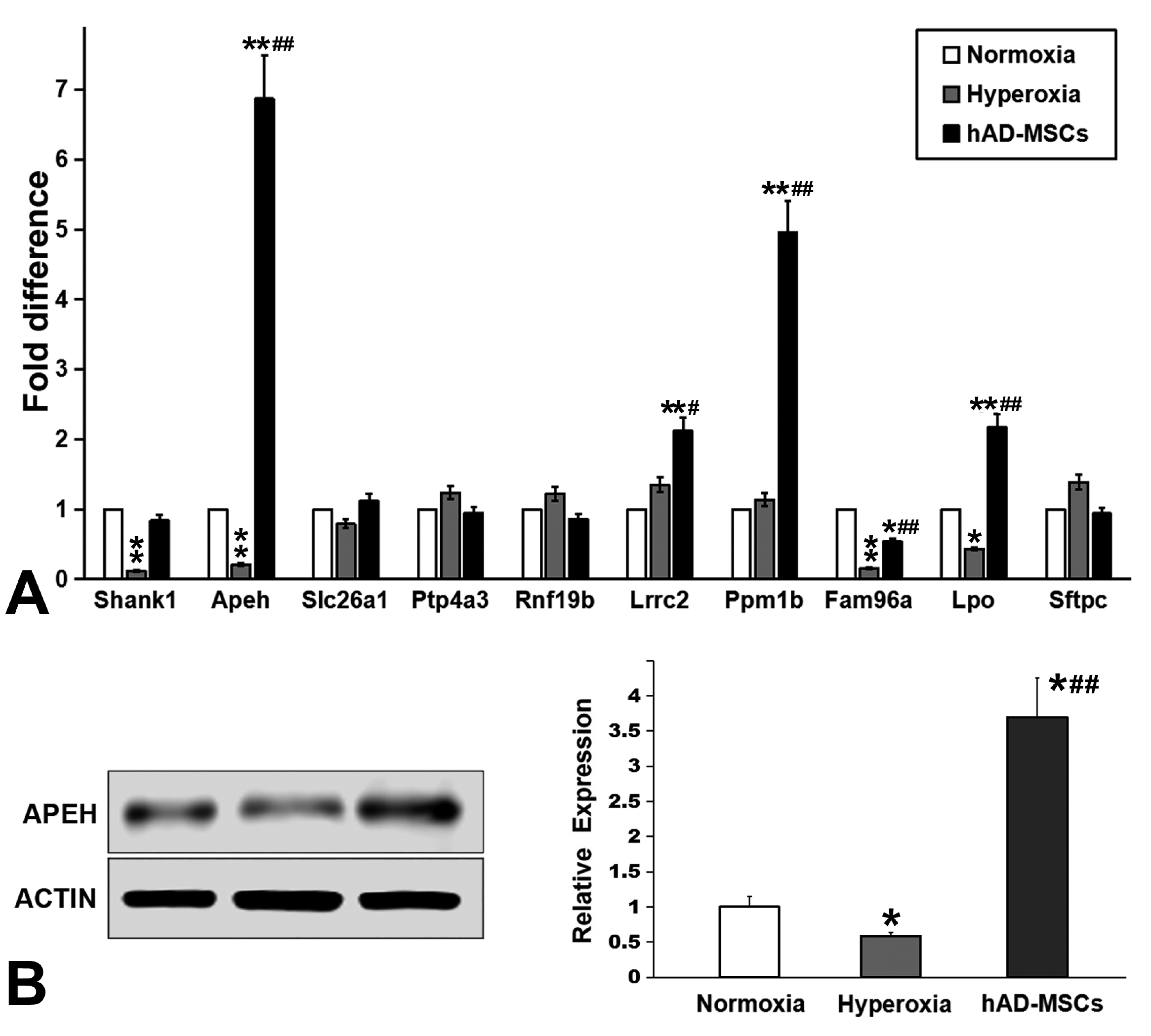 | Fig. 7The expression of APEH is closely related to lung injury and treatment with hAD-MSCs. (A) RT-qPCR analysis was performed to confirm the expression profiles obtained from gene chip analysis. (B) The expression of APEH was verified using western blotting. *p<0.05 vs. Normoxia group, **p<0.01 vs. Normoxia group, #p<0.05 vs. Hyperoxia group, ##p<0.01 vs. Hyperoxia group.
|
Go to :
Discussion
In this study, we cultured and isolated hAD-MSCs, and then compared the therapeutic effects of hAD-MSCs, hAD-MSC-CM, and hAD-MSC-EXOs on NHLI in rats. We systematically investigated the overall changes in body condition, lung index, and inflammatory index of newborn rats. An intratracheal drip of living cells was more effective for treating NHLI than cellular secretions or exosomes in terms of both ameliorating pulmonary edema and blocking inflammatory cell infiltration. And living cells therapy could better improve the survival rate of animals than secretome. This is primarily because living cells can survive in the lungs for several days and secrete greater concentrations factors and exosomes. Therefore, in subsequent gene analysis, we only evaluated rats in the hAD-MSC group, and underwent a comparative analysis of differential gene expression in lung tissues.
In line with these results, although conditioned medium (CM) contained cytokines including TGF-β, stromal cell-derived factor 1 (SDF-1), hepatocyte growth factor (HGF), monocyte chemotactic protein 1 (MCP-1), vascular endothelial growth factor-A (VEGF-A), and insulin-like growth factor-1 (IGF-1) (15), the therapeutic effect of CM remained limited (16). For instance, MSCs were previously shown to ameliorate fulminant hepatic failure and chronic liver fibrosis in mice, while MSC-CM could only partially ameliorate hepatic failure (17). In the treatment of acute kidney injury (AKI), although MSC infusion can improve renal function and significantly reduce mortality. However, treatment with CM showed a non-significant improvement (18). Although cytokines and nucleic acids play a major role in MSC-CM, they are clearly affected by proteases and nucleases in the microenvironment of the body and can only play a short-term role before their degradation. Previous findings showed that the low concentrations of growth factors in non-activated MSC-CM also hampered its clinical application (19). In contrast, MSCs were shown to survive for several days longer in vivo and play a sustained role. After MSCs were i.v. infused into mice, most of the cells were trapped in lung and disappeared with a half-life of about 24 hr (20). Nonhuman primate studies demonstrated that 7 days after infusion of allogeneic or autologous MSCs, only a minor fraction of the cells (less than 3%) engraft in different tissues (21); the majority of these cells were found in the kidney, lung, thymus, and skin (22). Recent findings showed that a very low concentration of MSCs was identified in vivo after 4 weeks (23).
Although the immunoregulatory role of MSCs in vitro and in vivo, they also clearly demonstrate that MSCs cannot completely evade the immune system and are eventually rejected (24, 25). Whether MSCs died of immune rejection or inflammation, the benefits of secretion of exosomes and improvement of microenvironment will continue for a period of time. MSCs-derived exosomes provide a protective membrane to protect cytokines and nucleic acids from enzymatic degradation during transport. These small vesicles are widely involved in intercellular communication and can alter the metabolism of target cells or local tissue microenvironment. Recently, MSC-derived exosomes were shown to mediate the therapeutic efficacy of MSCs in various disorders, such as acute kidney injury (26), cardiovascular disease (27), lung injury (28), radiation-induced hematopoietic failure (29), and liver diseases (30). However, high amounts of MSC-CM are required to obtain a small concentration of exosomes. Furthermore, exosomes can only play a one-off role, which is therefore less durable than the continuous production of exosomes by living cells.
In the treatment of BPD, some studies have shown that the intravenous (IT) hMSC administration route is as effective as intratracheal (IV) administrations (8, 9). Clinical studies investigating the appropriate dose of IT MSCs for treatment of BPD included doses of 1×107 and 2×107 cells/kg. Both doses appeared to be safe without increased short term or long-term adverse events (31, 32). Multiple studies have investigated the efficacy of IT MSCs delivery in rat BPD model, and these have typically included doses of ∼105 cells per rat (33, 34). But we found in the pre experiment that a relatively high MSCs IT dose (1×106 per rat) resulted in greater animal survival, which showed maximal efficacy as well as favorable safety with this dose.
Many kinds of proinflammatory cytokines are activated during oxidative stress, such as IL-1β, IL-6, MCP-1 and TNF-α (3, 35, 36). High expression of these factors promotes chronic inflammation, leading to the development of BPD. Experiments in neonatal rats have shown that the inhibition of inflammatory factors is beneficial for treating alveolar and lung injury by reducing lung inflammation and oxidative stress. For example, the inhibition of TNF-α can decrease the levels of MDA, which is helpful for lung development and pulmonary vascularization (37). IL-1β, IL-6, and MCP-1 are also identified as the biomarkers in monitoring ALI (35, 36, 38). In this study, we found that transfusion with hAD-MSCs successfully inhibited the expression of these proinflammatory cytokines in lung tissue. These reduction effects of human MSCs on proinflammatory cytokines are consistent with relevant studies (39, 40). These results suggested that the therapeutic effects of MSCs on developing lungs are partially mediated through the inhibition of proinflammatory cytokine production.
These inflammatory microenvironment also affects MSCs. The short-term, low-level exposure with TLR4 agonists polarizes hMSCs toward a pro-inflammatory MSC1 phenotype important for early injury responses (41, 42). High level of proinflammatory cytokine may promote anti-inflammatory MSC2, and induce decreased inflammation (43). In addition, hyperoxia augmented proinflammatory M1 macrophage phenotype and inhibited M2 polarization in macrophages (44). However, various tissue-derived MSCs, human adipose tissue-derived MSCs (45), BM-MSCs (46), and amniotic MSCs (47) could induce immunomodulatory M2-like macrophages. These findings revealed that MSCs may suppress inflammatory response and alleviate injuries in BPD mice via M1/M2 macrophages switch (48). Although MSCs administration can increase the pool of M2 macrophage and Tregs, some long term follow up studies (>6 months after intervention) suggest that the beneficial effects of the MSC-therapy wear off with time and may necessitate repeat treatment (49).
Although the mechanism of MSCs in the therapy of NHLI has been explored by many researchers, few studies have focused on significantly differentially expressed genes in lung cells after MSC treatment. Through gene chip hybridization, PCR, and western blotting, we showed that APEH expression was significantly decreased after hyperoxia injury, and increased significantly after hAD-MSC treatment. APEH catalyzes the removal of N-acylated amino acids from acetylated peptides, and has been postulated to play a key role in protein degradation. This proteasome modulates protein homeostasis and controls the turnover of regulatory proteins involved in critical cellular processes, including cell cycle progression and apoptosis (50).
In the process of NHLI, oxidative stress and oxidative damage lead to major lung injury (51). Neutrophils in lung tissue are the main source of reactive oxygen species (ROS). Malondialdehyde (MDA) is the main product of lipid peroxidation. In the assessment of body injury, MDA is mostly used to assess the degree of oxidative damage (52). Our results showed that the intrahracheal transplantation of hAD-MSCs significantly reduced MDA content in NHLI lung tissue, indicating that the redox environment was improved. Recent studies have shown that premature exposure to hyperoxia and the role of ROS are the main risk factors for BPD (53). ROS are highly active oxidants that directly damage cellular structures as well as individual proteins, lipids, and nucleic acids (54, 55). ROS can initiate the oxidation and nitrosylation of proteins, resulting in effects such as carbonylation, nitration, and nitrotyrosine formation, and ultimately leading to major cellular dysfunction (56). Cells have developed a whole defense system against ROS, including SOD, catalase and glutathione peroxidase (57). In addition to these enzymes, APEH is another ubiquitous cytosolic protease, which plays an important role in the detoxification of oxidized proteins.
In mammalian cells, APEH has been found to pay an important role in the clearance of oxidatively damaged proteins, as well as in the degradation of β-amyloid peptides in Alzheimer’s disease (58). Zeng et al. (59) also identified APEH as an integral part of the cellular response to DNA damage. APEH is helpful to repair single-strand DNA breaks after cell injury, and to increase the survival rate of cells exposed to ROS. The infusion of hAD-MSCs into neonatal rats with hyperoxia injury significantly reduced ROS production and MDA levels in this study. This suggests that hAD-MSC could significantly inhibit the production of ROS and reduce the oxidative damage in lung tissue. This effect was shown to be potentially closely related to increased APEH production.
In summary, the intratracheal application of hAD-MSC ameliorated the symptoms of NHLI in neonatal rats, with APEH playing a major role in this process. Understanding the relationship between APEH expression and lung injury may provide a theoretical basis for the clinical diagnosis and treatment of neonatal lung diseases. For example, measuring changes in APEH expression after pregnancy may help to quantify the degree of lung injury. Interventions targeting APEH can potentially improve the long-term quality of life of premature infants with lung injuries.
Go to :
 XML Download
XML Download