

 PDF
PDF Citation
Citation Print
Print


Introduction
Mesenchymal stem cells (MSCs) are multipotent cells which can produce a variety of differentiated cell/tissue lineages, including cartilage, bone, adipose tissue, tendons, and ligaments (1-3). MSCs can be recruited to sites of inflammation and tissue repair. Therefore, human MSCs (hMSCs) that maintain their capacity for self-renewal ability contribute to a wide variety of endogenous organ and tissue repair. In this rapidly expanding field, more and more attention has been paid to the relationship between stem cells and tumor cells. Much research found that MSCs tend to migrate to the tumor microenvironment, which has led to increased interest in using MSCs as carriers to deliver anti-tumor drugs or genes for cancer treatment. However, the nontherapeutic MSCs play a key role in cancer progression or treatment which is still without a conclusion (4).
Numbers of studies demonstrated that MSCs can promote tumor growth or metastasis and even related to the formation of the tumor-supporting stroma. More specifically, it has been reported that nontherapeutic MSCs enhanced tumor growth on HCC cells in vivo (5). Similarly, Zhu et al. (6) showed that MSCs could enhance the invasive capacity of cancer cells via extensive angiogenesis and tumor cell protection of immune cell recognition. However, growing evidence shows that hMSCs home to sites of tumorigenesis, where they exhibit antitumor effects both in vitro and in different cancer mice models. For example, hMSCs are recruited with high tumor specificity to gliomas in the brain, and they prolong the survival of tumor-bearing animals (7). Kidd et al. (8) observed that in an in vivo model of pancreatic cancer, intraperitoneally injected hMSCs migrated to primary and metastatic tumor sites and potentially inhibited tumor growth. Maestroni et al. (9) also showed that co-injection of mice MSCs with tumor cells can decrease the tumor volume. In addition, the stem cell microenvironment plays an important role in preventing carcinogenesis by providing signals that inhibit cell proliferation and stimulate differentiation (10-12). Thus, MSCs can have therapeutic effects even if MSCs are not transplanted or differentiated into cells of a particular problem, which could significantly increase the range of MSC therapeutic applications (13).
MSCs therapy received more attention from researchers in hopes of revealing clues about their therapeutic efficiency. During in vivo condition, it is difficult to dynamics tracking the efficiency of the MSCs therapy (14). Most researchers use genetic markers such as Y-chromosome when male cells are introduced into females or fluorescent protein reporter genes, but these methods do not resolve the dynamics of cellular and temporal responses and are not quantitative (14). Noninvasive in vivo imaging accomplished by using bioluminescence imaging (BLI) can be a possible solution. BLI is a powerful methodology that has been developed over the last decade as a tool for molecular imaging of small laboratory animals, enabling the study of ongoing biological processes in vivo (15, 16). The principle of BLI as a valuable tool for tracking cell populations in vivo by measuring light emitted from cells that express light-generating enzymes, such as firefly luciferase. The main advantage of BLI is that even at very low levels of signal, as few as 100 cells can be detected in vivo. This form of optical imaging is low cost and facilitates real-time analysis of disease processes at the molecular level in living organisms (17, 18). BLI has been a valuable tool for use in longitudinal assessment of transplanted stem cell fate both in vitro and in vivo, by labeling cells with a constitutively expressing bioluminescent reporter gene. In addition, some people use them to track the immune cells and bacteria (19, 20). Currently, BLI has also been used in cancer research to track metastasis and measure tumor burden by using luciferase-expressing tumor cells. Injection of HeLa cells labeled with luciferase into the tail veins of severe combined immunodeficient mice allowed visualization of lung metastasis.
Bone marrow-derived MSCs (BM-MSCs) are the most common cell source, especially in animal-based experiments, for tissue repair, engineering, and vehicles for cell-based gene therapy (21, 22). However, the clinical application of BM-MSCs is limited due to the invasive nature of the sample collection, low cell yield, reduced proliferation, and differentiation capacities in aging donors, and some existing ethical concerns (23). Unlike BM-MSCs, human umbilical cord-derived MSCs (UC-MSCs) are viewed as a better choice of MSCs for clinical application due to the painless collection procedure, high cell vitality, low immunogenicity, the high paracrine potential for accelerating injury tissue repair processes, and the absence of ethical issues. Moreover, UC-MSCs have also attracted attention due to their immunomodulatory properties (23, 24). Today, UC-MSCs are being proposed as a possible multifunctional tool for regenerative medicine and immunotherapy.
Hepatocellular carcinoma (HCC), the primary malignant tumor of the liver, has become the third leading cause of cancer death worldwide, with more than 500,000 diagnosed each year (25). HCC occurs primarily in patients with underlying chronic liver disease and cirrhosis with various molecular causes, local expansion, intrahepatic spread, and ultimately distant metastatic progression (26). Despite the existence of many cancer treatments, including curative surgical resection, chemotherapy, radiation therapy, liver transplantation and radiofrequency ablation, these therapeutic effects are still not particularly desirable (27-29). Herein, the study aimed to utilize the BLI technology to confirm the effect of UC-MSCs on inhibiting the growth of liver cancer cells in vivo. To conduct this study, we first labeled the liver cancer cells H7402 with CMV-Luc2-mKate2 which can express Luc2 and mKate 2 (30). Then the efficiency of therapy was confirmed by treating with conditioned media and co-culture with cells. Finally, in vivo BLI was conducted to track in a time-dependent manner, as well as lesion size and histological changes, provides a role for UC-MSCs in the treatment of cancer.
Materials and Methods
Male Balb/c nude mice 4∼6 weeks of age were purchased from the center of the animal in Zhengzhou University. The animals were maintained in polycarbonate cages, with a dedicated aseptic environment. During the experimental period, all of the research protocols were approved by the biomedical research ethics committee of the Faculty of Medicine, Zhengzhou University. All the institution guidelines for conducting animal research were followed throughout this work.
Extraction and culture of UC-MSCs
Human mesenchymal stem cells were isolated from umbilical cord Wharton’s Jelly. The isolation of UC-MSC was performed according to the protocol previously reported (31). In brief, the umbilical cord was cut into 2 cm pieces and opened with a scalpel. The Wharton’s Jelly was scratched out and the vessels were removed. Thereafter, the Wharton’s Jelly was digested for 24 h in collagenase/DMEM solution (172 I.U./ml, Sigma, Steinheim, Germany) and the enzyme reaction was stopped by the addition of serum containing stem cell medium. After centrifugation for 10 min (500 g), the cell pellet was resuspended and incubated in trypsin (2.5%, Sigma, Steinheim, Germany) at 37℃ for 30 min. Again, the enzyme reaction was stopped by the addition of medium and centrifugation. Finally, the cell pellet was resuspended in stem cell medium and cells were seeded in a T75 culture flask. Non-adherent cells were removed by initial medium change after 24 h and media were replaced 3 days after initial plating.
The osteogenic, adipogenic, and chondrogenic differentiation identified by Alizarin Red staining, Oil Red O staining, and Alcian Blue staining, respectively. To accomplish the differentiation, passages 4 MSCs were cultured in OriCellTM UC-MSCs osteogenic, adipogenic, or chondrogenic differentiation media (Sigma Aldrich, St. Louis, MO, USA) as described by the manufacturer.
After the third passage, UC-MSCs were harvested by trypsin digestion. Resuspend 1×105 cells in 100 μl of cold FACS buffer per EP tube and were then stain with fluorescein isothiocyanate (FITC) or phycoerythrin (PE)-conjugated mouse anti-human monoclonal antibodies (CD 34, CD44, CD45, CD73, CD90, CD54, CD105, HLA-DR) (Invitrogen, Carlsbad, CA, USA) for incubation on ice 30 min in the dark. The expression of every antigen is shown together with their corresponding isotype control. After washing twice with FACS buffer, the cells were fixed with 1% paraformaldehyde (Sigma) in PBS. Cell fluorescence was evaluated by flow cytometry using a FACS Calibur instrument and the data analyzed using Cell Quest software (BD).
Culture and protein labeling of H7402 cells
H7402 cells were obtained from ATCC (American Type Culture Collection) and were grown in a sterile 75 cm2 tissue culture flask in complete medium containing DMEM supplemented with 10% FBS and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) in 5% CO2 at 37℃.
A self-inactivating HIV-1-based lentiviral vector encoding luciferase 2 (lentiviral vectors of pLenti-6.3/DEST-CMV-Luciferase2-mKate2 were conducted by Invitrogen Shanghai) was constructed using multisite gateway technology. A lentivirus carrying pLenti-6.3/DEST-CMV-luciferase2-mkate2 gene was produced by transfecting 293FT cells with the expression construct and a lentiviral packaging mix, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The lentivirus was concentrated by ultracentrifugation at 4℃ and stored at −80℃ prior to use. H7402 cells were transduced with above mentioned lentiviral particles using Lipofectamine reagent (Invitrogen Corporation, USA) according to the manufacturer’s protocol. After FACS sorting, most transduced cells stably expressed reporter gene CMV-Luciferase2-mKate2 and this cell line obtained was termed H7402-CMV-Luc2-mKate2.
Collection of conditioned media
UC-MSCs were cultured as described above to 100% confluence. The conditioned media from the UC-MSCs were harvested and stored at −80℃ until use.
Cell viability assay of H7402 treated with conditioned media
Briefly, H7402 cells (5×104 cells/well, 100 μl) were seeded into 96-well plates and allowed to attach overnight. H7402 were pretreated with RPMI 1640 medium containing 10% fetal calf serum, UC-MSCs-conditioned media and a mixed medium of both with a proportion (1:1 to 1:20). After incubation for 96 h, 10 μl of CCK-8 (Dojindo, Kumamoto, Japan) was added to each well, and the cells were further incubated away from light at 37℃ for one hour. Three wells were used for each group. Cell viability was determined from absorbance readings at 450 nm with a 96-well plate reader (DG5032, Huadong, Nanjing, China). Data were expressed as the percentage of viable cells as follows: relative viability (%)=[A450(treated)−A450(blank)]/[A450(control)−A450(blank)]×100%.
H7402 cells were cultured and treated with UC-MSCs-conditioned media with a series concentration for 96 h incubation. For annexin-V and propidium iodide detection, Cells were labeled with either Annexin-FITC (Apoptotest FITC, DakoCytomation), or Propidium iodide (Invitrogen) as per the manufacturer’s recommendation. Apoptotic and necrotic cells were quantified using a BD FACS Calibur (Beckton Dickinson) and Cell quest pro software (Beckton Dickinson).
Effect of UC-MSCs on H7402 cancer cells
To determine the effect of UC-MSCs on liver tumor cells, we cultured an equal number of UC-MSCs and H7402-CMV-Luc2-mKate2 cells for 3 days. Briefly, UC-MSCs (1×105) were pre-plated in six-well plates overnight. H7402-CMV-Luc2-mKate2 cells (1×105) were added to the wells. After 3 days of culture in RPMI1640 complete medium. The morphology and interaction between the two types of cells were observed under a field of view by a fluorescence microscope. In addition, semi-quantitative analysis of the viability of H7402-CMV-Luc2-mKate2 cells was performed by measuring the fluorescence intensity in the selected region.
Transwell migration assay of UC-MSCs
Cell migration was assessed using Falcon HTS FluoroBlok 24-well inserts (BD Biosciences, San Jose, CA) with 8 μm pores. UC-MSCs (2×104) suspended in 200 μl DMEM (Invitrogen) with 10% FBS were seeded in the upper well of the transwell chamber, while RPMI 1640 medium, H7402-conditioned media or 5×104 H7402 cells per well suspended in 600 μl RPMI 1640 medium were added to the lower chamber of 24-well plate. After incubation at 37℃ for 12 h, the cells that had passed through UC-MSCs the transwell filter were stained with Geisma and counted in eight non-overlapping fields under a light microscope at ×200 magnification.
Balb/c nude mice transplantation
We obtained Balb/c nude mice which were the ethical treatment of the animals from the Center of Experimental Animals, The first affiliated hospital of Zhengzhou University. Mice of age 4∼6 weeks were used and kept in pathogen-free conditions. Mice were subcutaneously injected the nape region with a mixture of equal numbers of H7402-CMV-Luc2-mKate2 cells and UC-MSCs suspended in 100 μl saline. Mice that treated only with H7402-CMV-Luc2-mKate2 cells was set as control.
Bioluminescent imaging
BLI was performed with an IVIS Imaging System (Xenogen) comprised of a highly sensitive, cooled CCD camera mounted in a light-tight camera box. Images and measurements of bioluminescent signals were acquired and analyzed using Living Image Software (Xenogen). For in vitro imaging, bioluminescent cells were added into DMEM in 48-well plates. The wells without cells were set as control. D-luciferin was added to each well at final concentration of 150 μg/ml, and the photon emission from each well was acquired 1 s.
For in vivo imaging, Balb/c nude mice inoculated with H7402-CMV-Luc2-mKate2 cells were injected with D-luciferin at 150 mg/kg at 10 min prior to imaging. The luciferase activity was detected in vivo by the IVIS imaging system, digitized and electronically displayed as a pseudo-color overlay onto a grayscale animal image. Image acquisition times were 1 s. Regions of interest (ROI) from displayed images were drawn around the tumor sites and quantified as photons/second (p/s) using the Living Image software (Xenogen). BLI was performed at different time points beginning at 3 days after transplant.
Immunohistochemistry
For immunohistochemistry, these tumour-bearing mice were divided randomly into breast cancer control group (n=7) and the UC-MSCs treatment group (n=7). For immunohistochemistry performed to detect the distribution of UC-MSCs, paraffin-embedded sections of tumors were deparaffinized and rehydrated, with endogenous peroxidases quenched with 0.3% H2O2 in methanol. UC-MSC were detected with goat anti-ffLUC (Promega) antibody at a final concentration of 20 A g/ml. To determine if UC-MSC were localized in the tumor parenchyma or associated with tumor vasculature, immunohistochemistry was performed with mouse anti-CD31 (Abcam) and goat anti-ffLUC as described above. UC-MSC were then counted in six high-power fields for three sections and scored as either parenchymal or vascular-associated. The cells were counted by two independent blinded reviewers.
Results
Characterization of hUC-MSCs
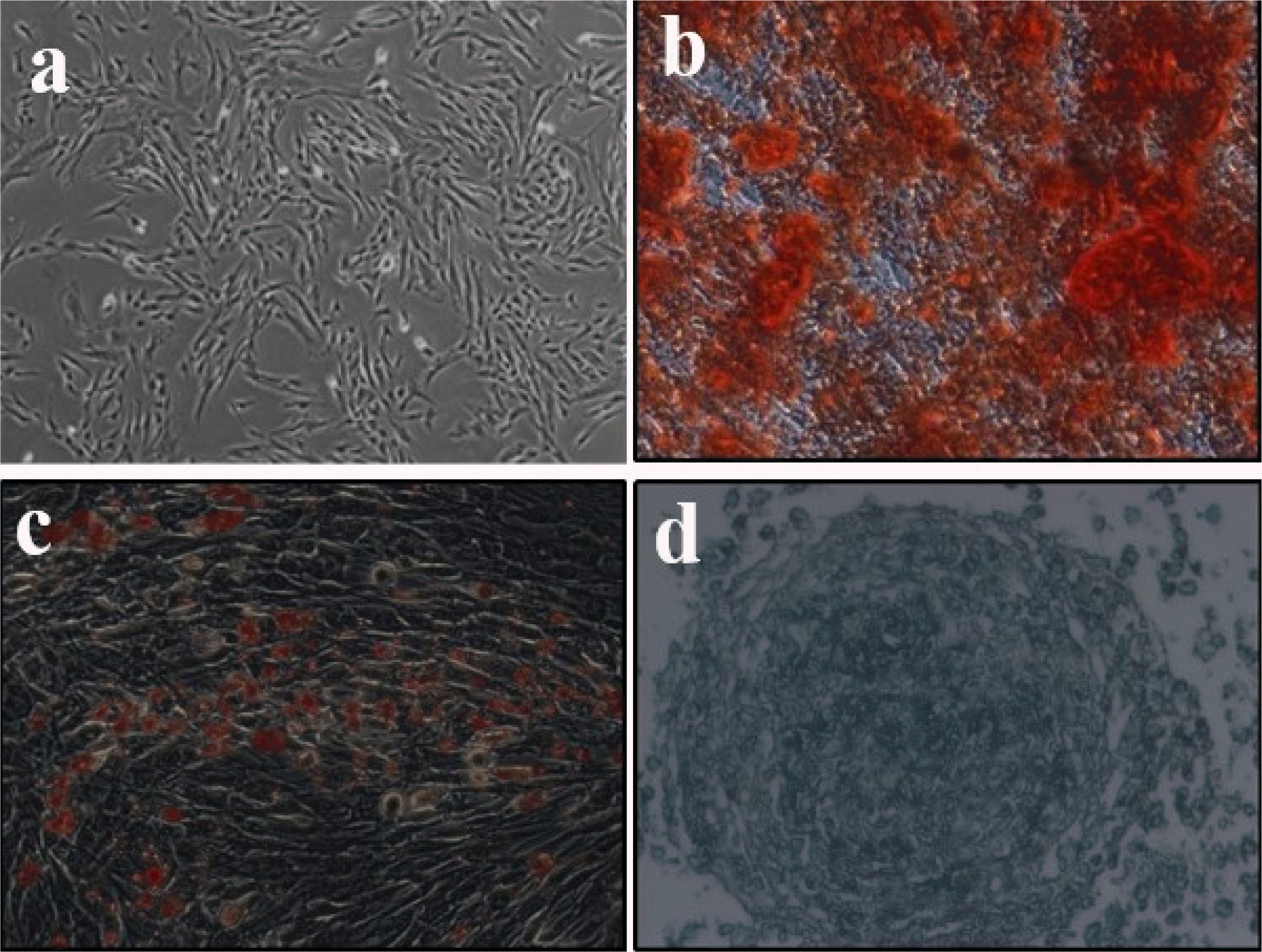
UC-MSCs exhibit typically fibroblast-like morphology (Fig. 1a) and flow cytometry analysis of surface marker expression revealed that these cells express high levels of CD 34, CD44, CD45, CD73, CD90, CD54, CD105, HLA-DR (Table 1), which is consistent with the characteristics of UC-MSCs that have been reported (32). UC-MSC has the potential to differentiate into osteogenic, adipogenic or chondrogenic in vitro under appropriate induction conditions. To verify the differentiation potential of UC-MSCs, cells were cultured with different induction medium. As shown in Fig. 1b∼d, mineralization deposition of osteogenesis was detected using alizarin red S staining, triacylglycerol accumulated in fat production was detected using oil red O staining, and cartilage-specific proteoglycan was detected using Alcian blue staining. All these data indicate that MSCs have pluripotent differentiation potential. Taken together, these findings indicate that cells display typical UC-MSC characteristics.
Generation of labeled tumor cells for real-time imaging
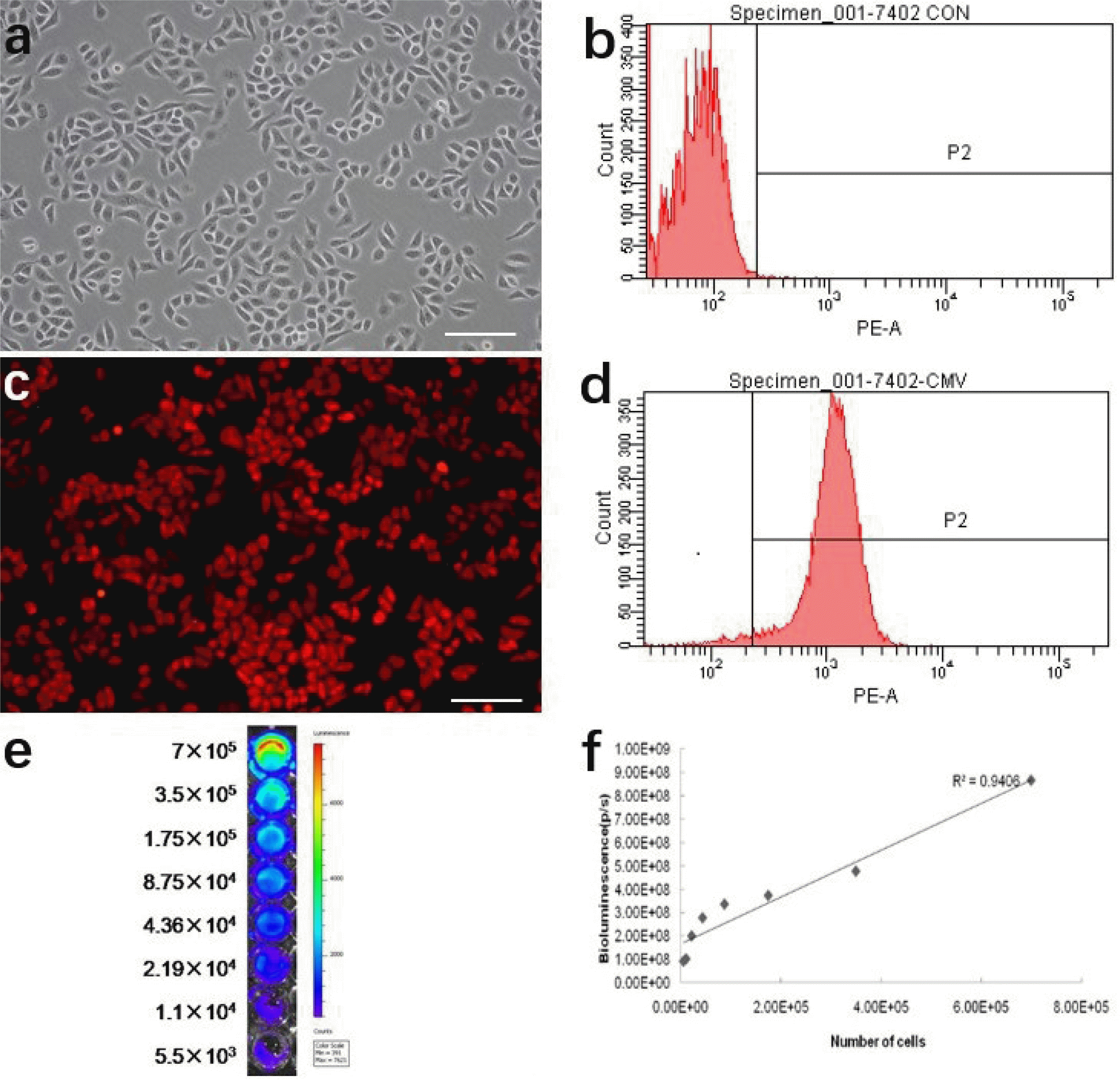
In order to track transplanted cancer cells in vivo using imaging analysis, we transduced H7402 cells with a pLenti-6.3/DEST-CMV-luciferase 2-mKate2 gene which stably expressed mKate2 and luciferase 2. Therefore, tracking cancer cell changes in vitro can be determined by imaging and analyzing the expression of mKate2 and luciferase 2 activity. As shown in Fig. 2a, we can see the H7402 cells adhesion growth under the bright field, which has a good morphology and growth state. Fluorescence imaging results indicated that mKate 2 was strongly expressed in H7402 cells and exhibited very bright red fluorescence (Fig. 2c). FACS analysis showed that the percentage of mKate 2 positive cells was 96.9% (Fig. 2d). In addition, we also found the strong signal of luciferase 2 in H7402 via bioluminescence imaging (Fig. 2e). we also can clearly see that as numbers of the cells increased, it exhibited a much stronger BLI signal. Moreover, a strong correlation between the activity of Luciferase 2 and the number of H7402 cells was observed in vitro using the Xenogen IVIS system (r2=0.9406), which revealed the availability of tumor growth in vivo by analyzing BLI signal intensity (Fig. 2f).
Proliferation of H7402 cells was inhibited by UC-MSCs conditioned media
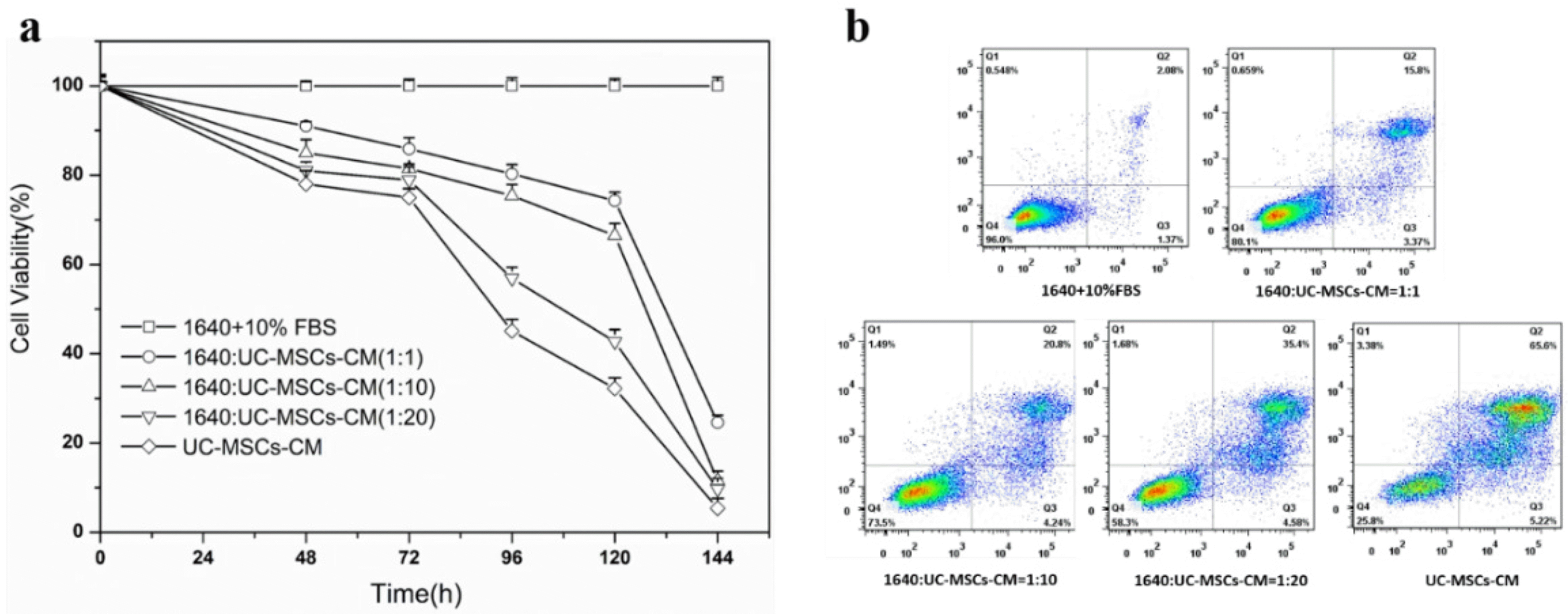
To investigate the effect of UC-MSC conditioned medium (UC-MSC-CM) on cancer cells, the viability of H7402 was assessed by CCK-8 after incubation with different levels of UC-MSC conditioned medium. As shown in Fig. 3a, the survival rate of H7402 cells treated with UC-MSCs was significantly reduced compared to the control group (1640+10% FBS). More specifically, when the percentage of UC-MSC-CM increased from 1:1 to total conditioned medium, cell viability showed varying degrees of decline at different time points. In addition, compared with the control group, with the increase of incubation time, the survival rate of liver cancer cells decreased significantly. After five days, the survival rate of all cells containing conditioned medium was less than 80%.
We further performed quantitative analysis to estimate apoptotic cell death by FACS. Our studies demonstrated that the apoptosis level of H7402 treated with UC-MSCs-CM for was increased that is consistent with CCK-8 assay. From the Fig. 3b, we can see that the survival rate of H7402 cells are 96% without treated with UC-MSCs-CM in 96 h, and as the concentration of the UC-MSCs-CM increased, there was an increase apoptotic rate of H7402 cells after treatment with complex media. After incubated the H7402 cells with the whole UC-MSCs-CM, the viability of cells even decreased to 25.8% after 4 days.
Effect of UC-MSCs on the proliferation of aggressive liver cancer cells
To study the influence of UC-MSC to the H7402-CMV-Luc2-mKate2 cells, H7402 cells were co-cultured with UC-MSCs. The growing status of tumor cells were observed by fluorescence imaging because of the express of luciferase 2. As shown in Supplementary Fig. S1a, the extent of H7402 cell death were dramatically increased after co-culture with UC-MSCs with typical morphological features of apoptosis include nuclear condensation and fragmentation form the bright field. By quantitative analysis of cell fluorescence intensity under field of view (Supplementary Fig. S1b), the fluorescent intensity of the H7402 is also obviously decreased to 60% after co-culture with UC-MSCs compared with control.
Migratory capacity of UC-MSCs in vitro
We evaluated the migratory nature of UC-MSCs towards H7402 cells in vitro (Fig. 4). UC-MSCs exposed to H7402 conditioned media (H7402-CM) or H7402 show an increase in migration compared to UC-MSCs exposed to RPMI 1640. Importantly, UC-MSCs exposed to H7402 conditioned media were found to possess significantly greater migratory capacity than exposed to H7402 (p<0.01). The number of migrated cells increased by 44% in UC-MSCs exposed to H7402 conditioned media and 49% in UC-MSCs exposed to H7402 by cell count compared to the control.
Dynamically and quantitatively tracking human H7402 tumorigenesis suppression by UC-MSCs in vivo using BLI
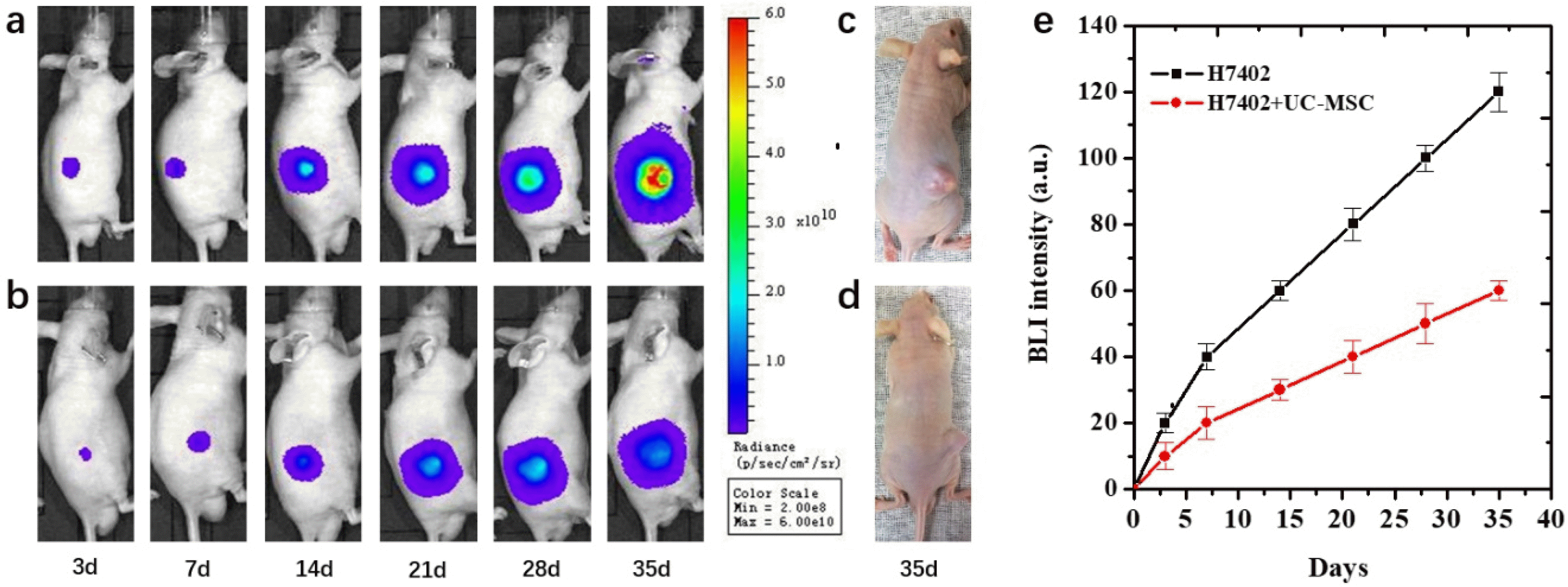
BLI was regularly performed beginning at 3 days after cells inoculation in two groups of mice with different experiments. The bioluminescence signal of mice with H7402-CMV-Luc2-mKate2 cells or complex of H7402-CMV-Luc2-mKate2 cells and UC-MSCs cells inoculation were shown in Fig. 5. The dynamic growth of the tumor was observed by BLI. At 3 days after cells inoculation, the bioluminescence signal of mice inoculated with complex of H7402-CMV-Luc2-mKate2 cells and UC-MSCs was significantly lower than those inoculated with only H7402-CMV-Luc2-mKate2 cells. The same tendency appeared in the following days, and after 35 days, a significant difference of the BLI signal between the co-injection group and the control group. Moreover, the inhibit efficacy of the UC-MCSs also can be clearly observed from Fig. 5c and 5d that the size of the tumor is much smaller than the untreated with UC-MCSs. In addition, from the quantitative analysis of the bioluminescence imaging signal in Fig. 5e, it was observed that the UC-MSCs cell treatment group showed a smaller imaging area and the bioluminescence intensity of treatment group also decreased about 54%.
Immunohistochemistry
Compared with liver cancer control group (n=7), tumor cells in UC-MSCs treatment group (n=7) were sparse and loose, with condensation of nuclei noted on haematoxylin and eosin staining. These findings demonstrate that the tumor cells were no longer largely proliferating and growing, and most of them went into apoptosis following UC-MSCs treatment.
Angiogenesis plays an essential role in cancer development; we therefore investigated the effect of MSCs on liver tumor microvessel density (MVD) that is an indicator of new blood vessel growth. Tumors from mice were evaluated by immunohistochemical staining (IHS) for the expression of an endothelial cell-specific marker CD31; and data in Fig. 6 show that the percentage of CD31-positive cells in control is much larger than MSCs-treated mice, suggesting that UC-MSCs caused inhibition of tumor growth is associated with its anti-angiogenic activity.
Discussion
HCC is the most common malignancy and is the leading cause of cancer-related death worldwide. At present, the main clinical therapy strategies include chemotherapy, surgery and radiotherapy have side effects (33-35). Mesenchymal stem cells (MSCs) are known to migrate to tumors, and it is possible to exploit the behavior of MSCs as a tumor-targeting method for cell-based cancer therapy. Numerous studies have investigated the effect of mesenchymal stem cells on different cancer types. However, the effects of MSCs on tumor progression remain controversial and no consistent conclusion has been presented until now. Most comments think that the efficiency of MSCs on cancer cells depending on the tumor type, MSC source, MSC dose, time of injection, study design, and the animal model. Bone marrow transplant is the most widely used stem-cell therapy, but recently some therapies derived from umbilical cord blood or human umbilical cord tissue mesenchymal cells (hUC-MSCs), isolated from fetal origins, have been studied for clinical use because UCMSCs are considered to be a more-primitive precursor. UC-MSCs have been found to target many primary solid tumors and their metastases. UC-MSCs secrete interferon-b (IFN-b), which was found to reduce the growth of human MDA-MB-231 breast carcinoma cells by inducing apoptosis. It was recently shown that the intratumoral injection of rat umbilical cord matrix stem cells (rUC-MSCs) caused regression of rat mammary carcinomas (36). Human umbilical cord Wharton’s jelly stem cells (hWJSCs) have been shown to have anti-inflammatory potential by reducing the expression of inflammatory mediators. Nevertheless, UCMSCs have been also reported to promote cancer growth and metastasis both in vivo and in vitro. The results concerning the effect of UCMSC on tumor growth were still without a conclusion. In the present study, UC-MSCs from human umbilical cord tissues possess an extensive potential to proliferate and differentiate into osteocytes, adipocytes, and chondrocytes. We demonstrated that these were homogeneously positive for the mesenchymal cell markers CD29, CD90 and CD44 but negative for CD34 and HLA-DR. These results are consistent with those of previous studies. The data showed that UC-MSCs were Alizarin Red-positive and Oil Red O-positive after induction. Taken together, the findings suggest that the isolated adherent cells from the umbilical cord were in fact MSCs.
To study the influence of stem cells on the cancer cells growth inhibition clearly, a necessary imaging technology should be used. Over the last decade, a variety of imaging technologies have been investigated as tools for cancer diagnosis and monitoring response to cancer therapies, such as MRI, CT, PET, BLI (37). Molecular imaging offers the potential for noninvasive assessment of therapeutic responses and real-time monitoring of tumor procession simultaneously. In addition, molecular imaging provides the possibility to visualize and monitor cellular and molecular processes, such as metabolism, biosynthesis, angiogenesis, cell proliferation, and apoptosis. Different works demonstrate the use of imaging to investigate the fate of the cancer cells in vitro or tumor in vivo. For example, someone uses fluorescent dyes to track the development of the tumor or using the MRI to monitor the fate of the stem cell transplantation by contrast agents (38, 39). However, all of these methods are not stable for real-time monitoring of cancer cells that is not because of the degradation of the fluorescent dyes, but also the cell proliferation will reduce the signal of imaging. To improve the efficiency of imaging, we evaluated the effect of UC-MSCs on cancer procession in nude mice using fluorescence and BLI dual imaging system. Herein, H7402 cancer cells used as a model to evaluate the effect of UC-MSCs in the process of interaction between each other. Furthermore, a pLenti-6.3/DEST-CMV-luciferase2-mKate2 gene was chosen to transduce into H7402 cells that can stability express luciferase2 and mKate2 respectively. mKate, as a fluorescent protein, can emission red fluorescence under laser that can be used for selective the labeled cells in vitro. In another hand, the expression of luciferase2 can be used for BLI in vivo. From the experimental results, the introduced cells are highly expressed, exhibiting very high red fluorescence in vitro, and can also imagine all labeled cancer cells in vitro using bioluminescence technology. From the experimental results, the introduced cells are highly expressed, exhibiting very high red fluorescence in vitro, and can also imagine all labeled cancer cells in vitro using bioluminescence technology. This evidence demonstrates the successful expression of this double-marker protein in cancer cells and lay a solid foundation for studying the effects of stem cells on cancer cells.
The following experiment clearly demonstrated the UC-MSCs played a critical antitumor role in vitro and in vivo. From the CCK-8 assay and the FSCF, it can be found the viability of cancer cells is an obvious decline when the concentration of the UC-MSCs conditioned medium is improved compared to the control groups, that means the substance inside the conditioned medium can affect the growth the cancer cells. The reason of this phenomenon is assessed by several researchers who conduct a very similar experiment. For example, Lv et al. (40) think the high concentrated ascorbic acid in the medium can be harm to the cancer cells. And that’s why when co-culture the UC-MSCs with cancer cells, the survival rate of the cancer cells shown significantly decrees. That can say the MSCs microenvironment strongly influences the growth of the cancer cells and play a key role in stem therapy and different stems have a different effect on the typed of cancer. Some people found that the stem has inhibited effect on the cancer cells, but others in the opposite, the stem can improve the growth of the cancer cells. Therefore, the effect of MSCs on the cancer cells usually depends on the two types of cells, there didn’t have a common conclusion. From the results, we also see that the UC-MSC also showed high migration ability towards cancer cells. It is known that systemically administered MSCs can migrate to the tumors or wounded tissues, and this ability can realize the possibility of the MSCs migrate to the tumor site for the relative therapy. Bioluminescence imaging was used to track H7402-luc2 cells in live mice and assessed stem cell inhibition of tumors for at least 5 weeks. Although the related inhibition mechanism needs further research. It can be seen from Fig. 5 that both the volume and BL intensity of the tumor were significantly decreased from 3 to 35 days and this phenomenon is much obvious after 14 days. In this process, we also observed the growth of the tumor in the control group is much faster than the group treated with stem cells by observing the tumor size changes. After 35 days, those transplanted with H7402 cells formed tumors with average tumor diameter about 11 mm. By contrast, mice injected with both H7402 cells and UC-MSCs formed detectable tumors with an average tumor diameter about 8 mm. On the other hand, tumor blood vessels are also an indicator of tumor growth. The histopathological experiment demonstrated blood vessels of the tumor tissue are suppressed (about 80% compared with the control group) which was consistent with the results of the quantitive BL intensity in 35 days. All these results demonstrated that BLI can be used to tracking the effect of UC-MSC on the tumor in vivo. In addition, UC-MSC plays a key role in decreasing cell proliferation and the growth of the tumor.
Noninvasive imaging technology is an important method to understand the interaction between the biological reactions. In the present study, bioluminescent and fluorescent double-labeled cells were achieved by transfection of designed genes. This is not only realized synchronous and quantitative tracking the tumor suppression via interaction with UC-MSCs but also can be significant for multimodal optical imaging of the interaction between organisms. Moreover, our findings provide evidence that both the UC-MSCSs and their conditional medium are capable of inhibiting growth, migration, metastasis, and angiogenesis of cancer cells. All these details illustrated the changing and controlling tumor microenvironment plays an important role in the process of tumor’s survival and reproduction which represent a research direction for cancer treatment. Further studies are required to confirm this relative mechanism.
Supplemental Materials
Supplementary data including one figure can be found with this article online at http://pdf.medrang.co.kr/paper/pdf/IJSC/IJSC-13-s19098.pdf.


 XML Download
XML Download