

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Medulloblastoma (MB) is the most common malignant brain tumor in the pediatric population. Traditionally, MB has been classified as average- or high-risk, depending on patient age, presence of metastasis, extent of postsurgical residual disease, and histology in clinical risk stratification;1 and treatment has been intensified in high-risk patients. With recent developments in genomic research, molecular studies on MB have been conducted, which have identified various distinct subgroups.2345 In a consensus conference in 2010, the MB Working Group agreed upon the existence of four main MB subgroups: WNT, SHH, Group 3, and Group 4.6 A subsequent consensus conference regarding risk stratification in the context of subgroups further refined the risk groups on MB in patients aged 3–17 years as follows: low-risk (LR) (> 90% survival), standard-risk (SR) (75–90% survival), high-risk (HR) (50–75% survival), and very-high-risk (VHR) (< 50% survival).7
The diagnostic criteria of the four molecular subgroups in MB were defined in 2013,8 and the suggested algorithm of subgrouping was largely based on methylation or gene expression profiling, especially in Groups 3 and 4. However, methylation profiling and genome-wide expression profiling studies are not readily available in the clinical setting, and limited gene expression studies are commonly used in many institutions after the study by Northcott et al.9 Additionally, molecular studies that can confirm DNA aberrations such as TP53 mutation, MYC amplification, and chromosome 11 loss are also needed for risk classification. DNA panel sequencing can be readily used to detect these DNA aberrations and can also be used to support molecular subgroup classification.
In this study, we used limited gene expression studies and DNA panel sequencing for integrated diagnosis of MB, and investigated the effect of integrated diagnosis on the risk stratification of childhood MB by comparing the risk groups between the clinical and new risk stratifications.
METHODS
Patients
Patients aged 3–17 years who were pathologically diagnosed with MB between 2005 and 2021 and had archival tumor tissues for molecular study were enrolled in this study. Patients aged < 3 years or ≥ 18 years were excluded from this study, as the consensus conference defined risk groups only in MB patients aged 3–17 years.
Limited gene expression study
Formalin-fixed, paraffin-embedded (FFPE) tissues were used, and all tumor specimens were reviewed by a pathologist to determine the percentage of viable tumors and their adequacy for molecular tests. For molecular subgrouping, limited gene expression profiling was performed using the nCounter® system (NanoString Technologies, Seattle, WA, USA) according to the methods proposed by Northcott et al. in 2012.9 Briefly, RNA was extracted from FFPE tissue using an RNeasy FFPE Kit (Qiagen, Germantown, MD, USA), according to the manufacturer’s instructions. A custom codeset comprising 22 MB subgroup-specific signature genes and three housekeeping genes was used. Raw data were collected and processed using nSolver™ analysis software. Classification of MB molecular subgroups and class prediction was performed as described by Northcott et al.9
DNA panel sequencing and molecular subgrouping using sequencing data
We used the targeted sequencing panel pipeline PedSCAN for data analysis, which was designed to cover 335 target genes at the Samsung Genome Institute (Supplementary Table 1). Paired-end reads were aligned to the human reference genome (GRCh37/hg19) using BWA-MEM v0.7.5, SAMTOOLS v0.1.18, GATK v3.1-1, and Picard v1.93 (http://picard.sourceforge.net). MuTect v1.1.4, Lofreq v0.6.1, and VarDict v1.06 software were used to detect single nucleotide variants. Pindel v0.2.5a4 was used to detect small insertions and deletions (indels) < 30 bp in size. Genetic alterations with a variant of allele frequency < 1%, total coverage depth < 50, or variant count < 4 were considered spurious variants and excluded. Sequencing errors were filtered using a machine-learning algorithm with features extracted from the SAM files. We calculated the mean read coverage of each exon and normalized it according to the coverage of the target regions in that sample to identify the somatic copy number alterations. This normalized read coverage was standardized by dividing it by the expected total coverage of a reference population. The expected coverage at each exon was obtained from the median read coverage at that exon across a set of normal individual samples. Furthermore, the amplitude of copy numbers was adjusted based on an accurate estimation of the tumor purity in the sample. Adjusted amplitudes of copy numbers > 4 and < 1 were considered as amplifications and deletions, respectively. Additionally, changes > 1 or < -1 in the Log2 scale of the adjusted copy number fold in the chromosomal arms were considered as a gain or loss, respectively.
Risk stratifications
In clinical risk stratification, clinicopathological variables, such as the presence of residual lesion, metastasis, and histology were used to categorize patients into average-risk or high-risk MB; and high-risk MB was defined as MB with metastatic disease, postoperative residual tumor > 1.5 cm2, or large cell/anaplastic histology. New risk stratification incorporating integrated diagnosis was performed according to the results of a consensus conference in 2015,7 and patients were categorized into four risk groups: LR, SR, HR, and VHR.
Statistical analysis
Event-free survival (EFS) was calculated from the date of diagnosis until the date of cancer progression or treatment-related mortality (TRM), and overall survival (OS) was calculated from the date of diagnosis until date of all-cause death. EFS and OS rates were estimated using the Kaplan-Meier method, and differences in survival curves were compared using the log-rank test.
Ethics statement
This study was approved by the Institutional Review Board (IRB) of the Samsung Medical Center (IRB No. 2015-11-053 and 2018-08-182). Since 2017, a molecular study was prospectively performed at the time of diagnosis after obtaining written informed consent from the parents or guardians of each patient. The requirement for informed consent was waived for patients diagnosed between 2005 and 2016 due to the retrospective nature of the study.
RESULTS
Patients’ characteristics
In total, 44 patients (25 men) were enrolled. Characteristics of patients were summarized in Table 1. The median age at diagnosis was 8.3 years (range, 3.3–16.8 years), and 14 (31.8%) and 19 (43.2%) patients had postoperative gross residual tumor size of > 1.5 cm2, and metastatic disease at initial diagnosis, respectively. Histologically, 36, 4, and 4 patients had classic, desmoplastic/nodular, and large cell/anaplastic histology. Collectively, 24 and 20 patients were classified as high- and average-risk, respectively, using the clinical risk stratification.
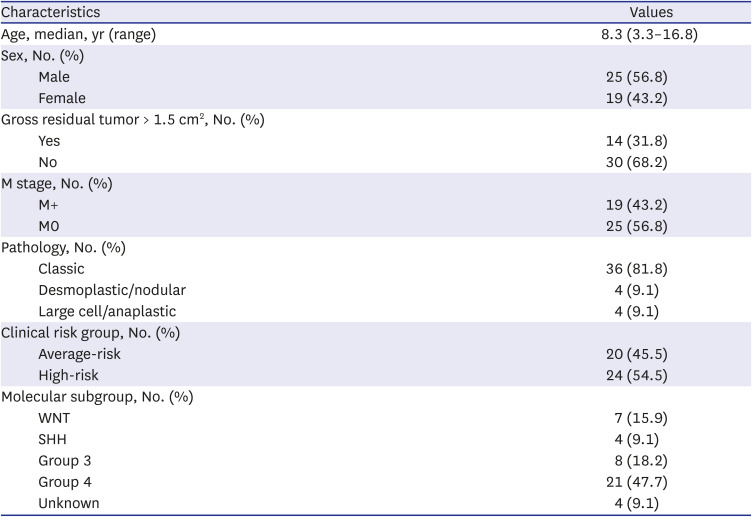
Table 1
Characteristics of patients
![]()
Molecular subgroups
Of the 44 patients, a limited gene expression study of 2 patients was not performed due to the quality standard criteria of RNA. Thus, limited gene expression studies were performed in the remaining 42 patients, and 37 patients were successfully classified into the WNT (n = 5), SHH (n = 3), Group 3 (n = 8), and Group 4 (n = 21). Five patients were unclassifiable with the limited gene expression study, as the results after class prediction analysis failed to fit any subtype.
DNA panel sequencing was successfully performed in all patients. Pathogenic variants of CTNNB1 were found in all five patients in the WNT subgroup, and monosomy 6 was detected in four of them. Among the three SHH patients, two had pathogenic variants of PTCH1 (PTCH1 deletion and PTCH1 E539*) and one had the TP53 V197G variant. This TP53 variant was confirmed as a germline variant and was classified as a likely pathogenic variant according to the ACMG guidelines. No MYC amplification was found in Group 3 patients, and one Group 4 patient had chromosome 11 loss.
Among seven patients whose samples were inadequate or unclassifiable in a limited gene expression study, two had pathogenic variants of CTNNB1 and monosomy 6. Additionally, one patient had a PTCH1 frameshift mutation. Combining the results of limited gene expression study and DNA panel sequencing, molecular classification was possible in 40 patients classified as follows: WNT (n = 7), SHH (n = 4), Group 3 (n = 8), and Group 4 (n = 21).
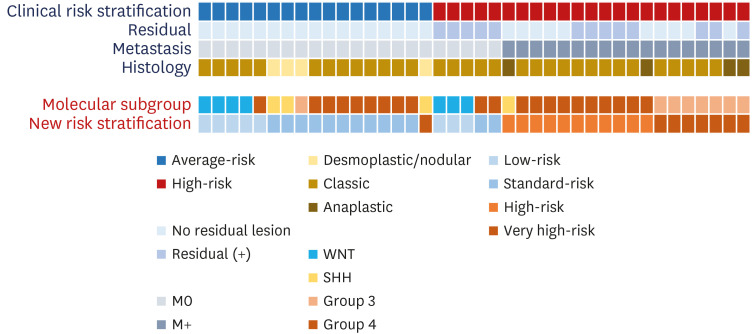
New risk stratification: discrepancy with clinical risk stratification
The 40 patients were stratified as follows using the new risk stratification: LR (n = 8), SR (n = 13), HR (n = 11), and VHR (n = 8) (Fig. 1). All seven WNT patients and one group 4 patient with loss of chromosome 11 were classified as LR; seven Group 3 patients with leptomeningeal seeding and one SHH patient with TP53 pathogenic variant were classified as VHR. When compared to the clinical risk stratification, among the 17 average-risk group patients, 16 patients were stratified into the LR or SR group. However, one patient in the SHH group with the TP53 variant who was classified into the average-risk group due to the absence of residual tumor and metastasis was reclassified into VHR. Eighteen of the 23 high-risk group patients were classified into the HR or VHR groups in the new risk stratification. However, five high-risk patients in the clinical risk stratification were classified into LR (n = 3) and SR (n = 2) groups in the new risk stratification. All five patients were classified as HR based on the clinical risk stratification due to residual tumor > 1.5 cm2 without metastasis or anaplastic histology. Three of them, with the WNT molecular subgroup, were classified into the LR group, and two with Group 4 were classified into the SR group.
Clinical outcomes
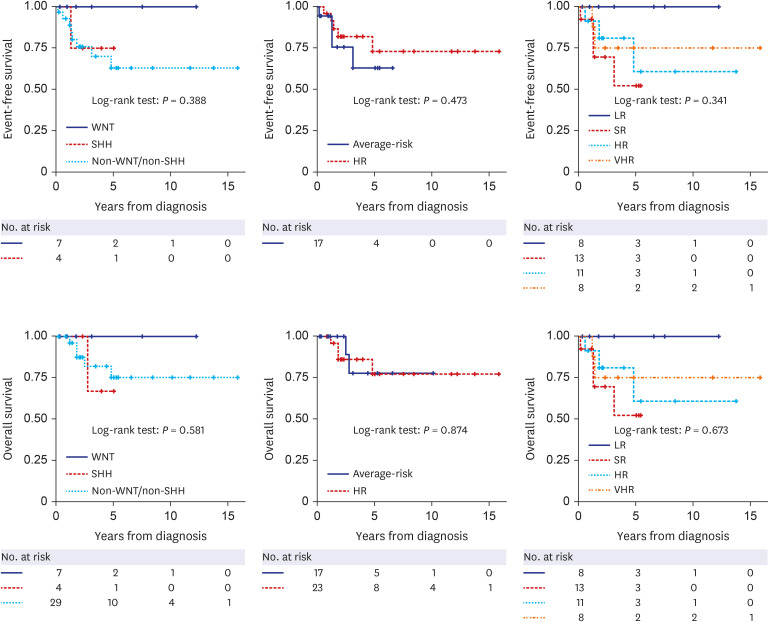
Surgery, two cycles of pre-radiotherapy (RT) chemotherapy, RT, and post-RT chemotherapy were performed in all patients, and tandem high-dose chemotherapy and autologous stem cell transplantation (HDCT/auto-SCT) were conducted in high-risk patients with clinical risk stratification as described previously.10 Of the 40 patients, six patients (SHH [n = 1], Group 3 [n = 1], and Group 4 [n = 4]) showed relapse/progression, and three (Group 3 [n = 1] and Group 4 [n = 2]) succumbed to TRM. The 5-year EFS and OS of all patients were 69.2 ± 9.1% and 77.0 ± 8.6%.
The EFS and OS according to molecular subgroup, clinical risk stratification, and new risk stratification are illustrated in Fig. 2. Patients in the WNT group showed 100% of EFS and OS, and the 5-year EFS and OS of the other molecular subgroups were 75.0 ± 21.7% and 66.7 ± 27.2% in SHH group and 62.9 ± 11.0% and 75.0 ± 10.0% in non-WNT/SHH group. There were no significant differences in EFS and OS according to the clinical risk stratification and the new risk stratification, however, the LR patients in the new risk stratification showed 100% EFS and OS.
DISCUSSION
We compared the results of new risk stratification, which integrates molecular subgroups determined by limited gene expression studies and DNA panel sequencing, with historical clinical risk stratification in MB. Using the new risk classification system, some patients’ risk groups were escalated (in 1 patient) or de-escalated (in 5 patients) compared to the clinical risk stratification.
Historically, MB was classified into standard/average-risk or high-risk groups based on the presence of residual tumor/metastasis and histology, and more intensive treatments, such as HDCT/auto-SCT or higher doses (36.0–39.6 Gy) of craniospinal irradiation were administered to patients with high-risk MB.1112131415 Our institution has also administered tandem HDCT/auto-SCT in high-risk patients,1016 and all the clinically high-risk patients in this study received tandem HDCT/auto-SCT. However, five out of 24 high-risk patients in the clinical risk stratification were reclassified into the LR or SR group; in particular, three WNT patients who were originally classified into the high-risk group due to residual tumor were reclassified into the LR group, according to the new risk stratification system. As patients with WNT-MB have an excellent prognosis, and since many clinical trials are exploring the possibility of deescalation of treatment in this group, it is imperative to correctly diagnose the molecular subgroups to administer a tailored treatment in patients with MB, considering the late effects of intensive treatment.17 This is also the case for SHH-MB with TP53 mutation, which is a VHR group in the new risk stratification, and hence can be classified into an average-risk group in clinical risk stratification resulting in insufficient treatment.
For new risk stratification, at least two molecular studies are needed, and molecular subgrouping is primarily based on either methylation profiling or gene expression profiling. However, methylation profiling or genome-wide gene expression profiling is not yet available in clinical settings. Therefore, we used a limited gene expression study for molecular subgrouping. DNA panel sequencing was used to identify DNA aberrations for risk stratification, and it could also be used for molecular subgrouping, especially in the case of WNT or SHH MB. In this study, molecular subgroups could be defined in an additional three out of seven patients whose samples were inadequate or unclassifiable in a limited gene expression study: two WNT patients with CTNNB1 mutation/monosomy 6 and 1 patient with PTCH1 frameshift mutation.
A problem with the new risk stratification system is that there are still many “unknown” categories that cannot be classified into specific risk groups, such as metastatic WNT-MB. Additionally, the new risk stratification system was applied only to patients aged 3–17 years. Moreover, the postoperative residual tumor > 1.5 cm2, which was a factor determining high-risk group in clinical risk stratification, was excluded from the new risk stratification. This was based on a study of MB patients from the Hospital for Sick Children in Toronto (n = 787), which demonstrated that near-total resection poses no additional survival risk compared to gross total resection.18 However, there are still some conflicting data about the meaning of subtotal resection,1920 and there could be a concern in de-escalating treatment intensity in this population. These unanswered questions need to be re-evaluated in future prospective clinical trials using a new risk stratification system.
In the survival analysis, patients with WNT-MB showed excellent prognosis. When applying the new risk stratification system, LR patients showed better survival outcomes than SR patients. However, the survival data in this study should be interpreted cautiously due to the retrospective nature of this study and the small number of patients in each group. The treatment scheme was different between the average-risk and high-risk patients, and an intensive treatment using tandem HDCT/auto-SCT could result in a similar survival rate in high-risk groups.
This study has some limitations in that the number of enrolled patients is small to cover all the various cases. Also, although the limited gene expression study is a practical method for clinical application, the molecular subgrouping was not possible in some patients. Furthermore, many recent reports have defined intra-subgroup heterogeneity showing different biological and clinical characteristics among the same subgroup, suggesting the application of enhanced molecularly guided risk stratification.2122 In order to adapt to the future changes, it would be necessary to implement standard methylation profiling study into clinical practice.
In conclusion, this study demonstrated that the new risk stratification incorporating integrated diagnosis is discordant with clinical risk stratification. Thus, precise molecular subgrouping and risk stratification using integrated diagnosis should be implemented in clinical practice for tailored treatment in patients with MB. Additionally, this new risk stratification will need to be evaluated in future clinical trials.
 XML Download
XML Download