

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The worldwide prevalence of liver cancer is 841,000 cases per year, making it the sixth most common cancer globally, and 782,000 deaths occur annually. With similar mortality to prevalence rates, hepatocellular carcinoma (HCC) ranks fifth in terms of global cases and second in terms of deaths in males [1]. HCC comprises 75-85% of liver cancer cases and has several known risk factors including chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections, alcohol abuse, autoimmune hepatitis, diabetes mellitus, obesity, and several metabolic diseases [2,3].
Genetic and epigenetic alterations that progressively accumulate in a background of chronic liver injury and inflammation lead to the initiation and progression of HCC, involving a multi-step process. The key events in the molecular pathogenesis of HCC were poorly understood until recent advances in sequencing technology enabled identification of the critical oncogenes and tumor suppressors. The National Institute of Health (NIH) launched The Cancer Genome Atlas (TCGA) project in 2005 to establish a coordinated teamscience effort to comprehensively characterize the molecular events in primary cancers and to provide these data to the public for use by researchers globally.
As part of the TCGA network, Wheeler et al. performed the first large-scale multi-platform analysis of HCC, including the evaluation of somatic mutations and DNA copy number alterations in 363 patients and of deoxyribonucleic acid (DNA) methylation, mRNA expression, microRNA (miRNA) expression, and protein expression in 196 patients, to understand the molecular landscape of HCCs [4]. This study was one of the most comprehensive integrative genomic analyses of HCC to date with clear delineation of the HCC genomic landscape. In parallel with the TCGA project, European researchers in basic, translational, and clinical fields of liver cancer launched the HEPTROMIC project to elucidate the cancer genome and subsequently optimize decisionmaking for liver cancer patients by identifying prognostic genomic determinants and oncogenic drivers [5].
This review, based on results from the projects mentioned above and recent findings, provides an overview of the genetic changes involved in development and progression of HCC. Since HCC is highly resistant to treatment, the translational clinical application of these new findings to identify a subset of patients who might respond to targeted and immunotherapies will be discussed.
Go to : 
LANDSCAPE OF GENOMIC ALTERATIONS
1. Somatic mutations and copy number alterations
Somatic mutations occur in the somatic (non-germ) cells and are inheritable. Somatic mutations in tumors are important as the transformation of a normal cell into a cancerous cell occurs sequentially through a few discrete genetic events [6]. Whole-exome sequencing led to the discovery of telomerase reverse transcriptase (TERT ) promoter mutations as the most common somatic mutation (40-65%) detected in HCC [4,7,8]. Telomerase is composed of RNA (telomerase RNA component, TERC ) an enzyme, as a rate-limiting component of the complex. TERT is required for telomere synthetase [9], and patients with a TERT promoter mutation experience telomere shortening, limiting the life span of human hepatocytes and thus, are associated with a higher risk of cirrhosis [10,11]. TCGA data analysis has shown that patients with the TERT promoter mutation were older, predominantly male, and more likely to be HCV positive than patients without the mutation. HBV insertion in the TERT promoter also occurs in 10-15% of HCCs [4,12,13]. The pre-malignant lesion in a cirrhotic liver exhibited the TERT promoter mutation in 6% of low-grade dysplastic nodules, 19% in high-grade dysplastic nodules, and increases dramatically in early HCCs (60%) [14,15]. TERT promoter mutation co-occurring with cyclin-dependent kinase inhibitor 2A (CDKN2A ) silencing by promoter hypermethylation is an early event which behaves as a “gatekeeper” during hepato-carcinogenesis [4,16].
The genomic landscape using the whole exome-sequencing of 363 HCC cases from the TCGA is shown in Fig. 1. The significantly mutated tumor suppressor gene, tumor protein P53 (TP53 ) (12-48%), is frequently present in advanced tumors. Other highly mutated suppressor genes were AXIN1 (5-15%) and CTNNB1 (11-37%). Both activate the WNT pathway and promote cell motility and proliferation [17]. The chromatin remodeling genes include AT-rich interaction domain 1A (ARID1A ) (4-17%), ARID2 (3-18%), and BRCA associated protein 1 (BAP1 ) (5%), which regulate transcription of genes. The loss of these tumor suppressor genes causes increased proliferation with poor prognosis [18-23]. Due to mutations, decreased albumin (ALB ) and apolipoprotein (APOB ) was observed in HCC relative to normal tissue in 13% and 10% of tumors, respectively, and may be associated with cancer-relevant metabolic pathways [4].
 | Figure 1.The landscape of genomic alterations in hepatocellular carcinomas. The top panel shows individual tumor mutation counts, and the middle panel shows clinical parameters. The bottom panel shows genes with significant levels of mutations (MutSig suite, FDR, <0.1) and mutation types are shown as legends at the bottom. Adapted from reference [4]. FDR, false discovery rate.
|
Somatic copy number alteration (SCNA) results in either the gain or loss of segments of genomic DNA. Copy number (CN) gain was most frequently detected in chromosomes 1q and 8q and CN loss in 8p and 17p. The driver oncogenes were cyclin D1 (CCND1 ) and fibroblast growth factor (FGF19 ) (11q13.3). Brivanib, an inhibitor of vascular endothelial growth factor (VEGF ) and FGF19 , did not elicit clinical benefits [24]. H3B-627, a potent inhibitor of fibroblast growth factor receptor 4 (FGFR4 ) and FGF19 driven HCC, is currently under phase 1 clinical trials and deserves further attention [25]. Other oncogenes were MYC (8q24.21), MET (7q31.2), VEGFA (6p21.1), and myeloid cell leukemia sequence 1 (MCL1 ) (1q21.3). TERT (5p15.33) was also amplified in 10% of HCC cases, while tumor suppressor genes such as RB1 (13q14.2) and CDKN2A (9p21.3) were prominent in HCC samples.
2. Epigenetic modification
Epigenetic information includes DNA modifications (including methylation or hydroxylation), histone composition changes, and post-translational modifications (including methylation, acetylation, and phosphorylation), chromatin remodeling, and microRNA and non-coding (nc) RNA (including long nc [lnc] RNA) expression changes [26]. Alterations of the epigenome have been correlated extensively with cancer development, progression, and resistance to therapy [27,28].
Gene silencing due to promoter hypermethylation is a hallmark of human cancer, as DNA methylation regulates cell differentiation and tumorigenesis [29]. Disproportionally enriched CDKN2A promoter hypermethylation leading to epigenetic silencing was observed in HCV positive tumors, which often co-occurred with TERT promoter and CTNNB1 mutations [4]. In HBV positive HCCs, HBV alters the epigenome via the HBV X (HBx) protein. HBx recruits DNA methyltransferase to the regulatory promoters of the tumor suppressor genes to silence their transcription by hypermethylation [30]. Other downregulated genes due to hypermethylation in HCCs include hedgehog interacting protein (HHIP ), a suppressor of Hedgehog signaling, a pathway important in hepato-carcinogenesis, carbamoyl phosphate synthase I (CPS1 ), a liver-specific rate-limiting enzyme of the urea cycle [4], adenomatous polyposis coli (APC ), and insulin-like growth factor 2 (IGF2 ) [28].
Besides DNA methylation, chromatin modification is another epigenetic mechanism of gene regulation in cancer cells [31] The commonly altered, well-studied chromatin modifiers in HCC are; upregulated enhancer of zeste 2 polycomb repressive complex 2 (EZH2 ), inactivating mutation of ARID1A (4-17%), and ARID2 (3-18%) [22,32,33]. EZH2 , a methyltransferase that mediates gene silencing via the trimethylation of H3K37 shows increased protein expression from the dysplastic nodule to early HCC and is positively correlated with poor survival [34]. ARID1A and ARID2 are proteins that belong to the switch/sucrose non-fermentable (SWI/SNF) ATPase-related chromatin-remodeling complex and have tumor suppressor functions [35]. However, a recent study showed that ARID1A is context-dependent, where the presence of ARID1A expression supports initial HCC development, but loss after tumor development enhances metastatic potential [36]. An interesting study by Hu et al. [37] shows a poor prognosis of ARID1A-deficient HCC mice dependent on increased angiopoietin 2 (Ang2 ) expression. In addition, ARID1A deficiency sensitizes tumors to Ang2 blockade by sorafenib treatment, indicating the possibility of ARID1A as a biomarker for anti-angiogenic therapy.
The transcribed genome in the form of RNAs that are not translated into proteins but with regulatory functions, are called ncRNAs. miRNAs are 22-nucleotide RNA molecules that are the most intensively studied ncRNAs. The best example of the recent epigenetic therapy of liver disease is with the miR-122 antagonist, the most frequent one accounting for 70% of the total miRNA [38]. miR-122 regulates genes in the cholesterol metabolism pathway, and a loss of miR-122 correlates with tumor size and invasiveness, making it an attractive therapeutic target for HCC intervention [39]. Other tumorsuppressive miRNAs that are silenced in HCC are miR-26, miR-199a, and miR-200a [40-42].
LncRNAs comprising 200-300 nucleotides have tissue-specific expression and are transcriptionally regulated by key tumor-suppressors or oncogenes [43,44]. Functionally, most of the known or predicted lncRNAs have not yet been characterized. Hepatocellular carcinoma up-regulated long noncoding RNA is considered the first lncRNA specifically upregulated in HCC [45]. HOX transcript antisense RNA also negatively regulates miR-218 expression in HCCs through EZH2 targeting miR-218-2 promoter, resulting in oncogenesis [46]. A recent study by Yang et al. [47] identified 917 recurrently deregulated lncRNAs that are correlated with the clinical data in a TCGA cohort and published liver cancer data, and lncRNAs HAND2-AS1 were found to be related to metastasis. The oncomirs that drive the progression of HCC are miR-21, miR-221, miR- 222, and miR-224, which are commonly upregulated in HCC [48,49]. The upregulated levels of miR-221/222 occur as an early event in tumor development and are the most highly expressed miRNAs in HCCs [50,51].
Go to :
MOLECULAR SUBTYPES
Integrative genomic analysis has facilitated the elucidation of the mutation landscape and pathways involved in the development and progression of HCC. Since HCCs at the same clinical stage differ significantly at the molecular level, the next step is to classify tumors based on the molecular events to improve the clinical management of HCC patients based on biomarkers.
In HCC, many molecular classifications have been reported based on genomic profiling. One pioneering study by Boyault et al. [52] elucidated the molecular diversity of HCC tumors using an unsupervised transcriptome-wide approach to classify many tumors. Boyault’s 16-gene signature classified HCCs into six main subtypes (G1-6), and each subtype showed distinctive characteristics (Fig. 2). G1-3 are characterized by chromosomal instability and lower survival rate compared to that of G4-6. HBV related tumors were classified into the G1 and G2 subgroups and were molecularly distinct from other HCCs.
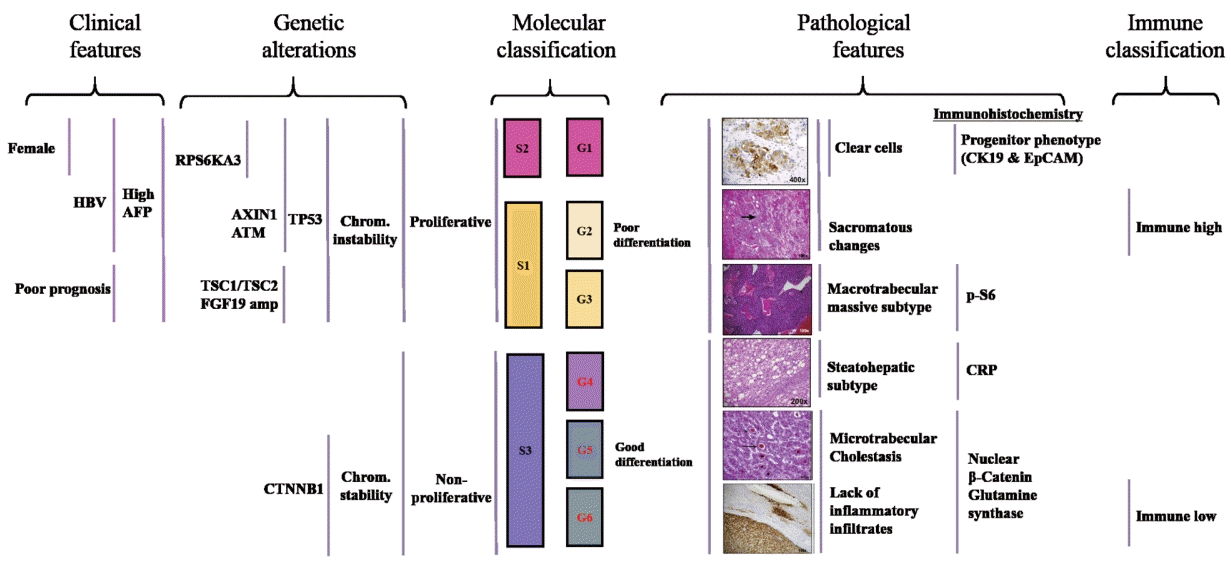 | Figure 2.Integration of molecular, pathological, clinical, and immune classifications. Proliferative classes (S1, S2, [Hoshida’s classification] and G1-3 [Boyault’s classification]) are associated with poor prognosis, and non-proliferative classes (S3, G4-6) are less aggressive subtypes. HBV, hepatitis B virus; AFP, alpha-fetoprotein; ATM, ataxia telangiectasia mutated; CK19, cytokeratin 19; CRP, C-reactive protein; CTNNB1, catenin beta 1; EpCAM, epithelial cell adhesion molecule; FGF19, fibroblast growth factor 19; p-S6, phospho-ribosomal protein S6; RPS6KA3, ribosomal protein S6 kinase A3; TSC1/2, tuberous sclerosis 1/2; TP53, tumor protein P53. Adapted from reference [54] with permission.
|
In contrast to tumors of HBV, HCV infection and alcohol abuse were classified across subgroups G3-6. Apart from HBV infection, low viral DNA copies, AXIN1 mutations, younger age, and high serum levels of α-fetoprotein (AFP) were observed in the G1 subtype, while high viral DNA copies and frequent local and vascular invasion with TP53 mutations were observed in G2. IGF2 overexpression in G1 and phosphatidylinositol 3-kinase catalytic subunit (PIK3CA ) mutations in G2 were predicted to activate the AKT pathway. The G3 subtype included tumors with the TP53 mutation and CDKN2A leading to cell cycle dysregulation and poor outcomes, while the G4 subtype was composed of mature hepatocytes (non-tumorous liver) without significant genetic alterations. The G5 and G6 subgroups were highly associated with β-catenin activation (70-100%) mainly due to CTNNB1 mutation and the inactivation of E-cadherin, which may be the cause of the local invasion of HCC.
Three common molecular subclasses named as S1, S2, and S3 were identified by Hoshida et al. [53] using the unsupervised clustering-based definition. Briefly, class S1 was associated with a higher risk of early recurrence and invasive/disseminative phenotype. This class showed a predominance of WNT pathway activation and interaction with transforming growth factor-β (TGF -β) activation. Class S2 tumors were associated with high levels of plasma AFP levels, MYC , and AKT activation signatures, and enrichment of positive EpCAM signatures. The TP53 mutation associated with stepwise malignant transformation was higher in the S1 and S2 classes than in the S3 class, while the β-catenin mutation was more common in well-differentiated S3 tumors. The authors recommended targeted agents such as β-catenin and PI3K inhibitors according to the molecular classifications.
Go to :
HISTOLOGICAL SUBTYPES
Based on a comprehensive overview of the molecular features mentioned above, the relationship between HCC molecular features and their phenotypes was determined in resected tumors by combining pathological analysis, gene expression profiling, and gene sequencing [54]. Two distinct HCC mutually exclusive phenotypes, delineated by CTNNB1 (40%) and TP53 (21%) mutations, were noted.
CTNNB1 -mutated tumors were large, well differentiated, cholestatic, and had micro-trabecular and pseudo-glandular patterns, and an absence of inflammatory infiltrates. In contrast, TP53 -mutated tumors were poorly differentiated with a compact pattern, multinucleated and pleomorphic cells, and frequent vascular invasion. In addition, TP53 -mutated tumors exhibited PI3K/AKT pathway activation as assessed by phospho-S6 protein in tumor cells and were associated with genes that showed increased cell proliferation, epithelial to mesenchymal transition, and angiogenesis activation. TP53 also strongly correlated with a novel subtype named “macrotubular-massive” (MTM), which was associated with poor survival, high AFP, vascular invasion, more frequent HBV infected tumor and increased FGF19 amplifications. Higher ANGPT2 mRNA levels, which is known to promote neo-angiogenesis and endothelial sprouting in cooperation of VEGFA , was also observed in MTM-HCC. This subtype was associated with the G3 subgroup (Fig. 2).
Another phenotype scirrhous HCC subtype, defined by marked stromal fibrosis, showed CK19K expression and the upregulation of progenitor/cancer stem cell genes (CD24 , KRT19 , THY1 , and CD133 ), and epithelial-to-mesenchymal transition activation (TGF -β, VIM ). The “Steatohepatitis ” subtype did not have specific clinical features but displayed a less aggressive phenotype. At the molecular level, this subtype correlated with the G4 subgroup. Major genetic alterations, according to tumor subtypes are shown in Fig. 2.
Based on the integrative analysis, highly reproducible HCC subtypes were developed (Fig. 2) [53,54]. Chromosomal instability, the G1-3 subgroups, S1, and S2 subtypes, and the proliferative subtypes are collectively called the “proliferative” subtype and are associated with more progressive tumors. In contrast, chromosomal stability, the G4-6 subgroups, and the S3 subtypes are called the “non-proliferative” subtype, with hepatic physiology resembling that of healthy individuals.
Go to :
CLINICAL APPLICATION FOR PRECISION CANCER MEDICINE
Genomic profiling enables the management of treatment for cancer patients by selecting appropriate targeted therapy based on tumor genotypes or other features. The “oncogene addiction theory” postulates that some cancers depend on one or a few genes for the maintenance of the malignant phenotype [55]. The successful translation of this principle in cancer management is evidenced by the treatment of HER-2/NEU positive breast cancer patients with the trastuzumab antibody targeting the receptor tyrosine kinase [56], and in BRAF V600E mutated melanoma patients treated with the vemurafenib antibody [57].
An appropriate biomarker should be established for the accurate identification of patient tumors expressing oncogene addiction to achieve survival benefits after selective inhibition. Challenges lie in developing targeted therapies for HCC, as the frequently mutated genes such as TERT , TP53 , CTNNB1 , and MYC are undruggable. Therefore, patients and clinicians had a long wait until promising results were reported for newly developed kinase inhibitors and immunotherapies. Despite significant improvements in the management of advanced HCC and promising results with novel therapies, a considerable number of patients (>60%) do not respond to these novel therapies [58,59]. The uncertainty of the combination of immune cells forming the immune microenvironment and the extent of intra-tumoral heterogeneity in HCC are the limiting factors for deriving predictive biomarkers to enhance responses to these novel therapies. With more options for sequential systemic treatments for advanced HCC, clinicians have to choose between sorafenib and lenvatinib as the first-line treatment and between regorafenib and nivolumab as the second-line treatment after sorafenib. Biomarkers research is a paramount research area, and the next section discusses predictive gene signatures and biomarkers discovered using the extensive exploratory biomarker analysis of DNA, RNA, and protein levels to identify a subset of patients who are likely to benefit from specific targeted therapies.
1. Immune features of HCCs
Sia et al. [60] identified an immune specific subtype of HCC by analyzing gene expression data from 228 HCCs using a non-negative matrix factorization algorithm to distinguish tumor, stromal, and immune cell signatures and validated these in 728 tumor samples. Using the gene signature, “immune class” that comprises 24% of total patients were identified, and this group captured the 1) signatures of immune cells (i.e., T-cells, cytotoxic cells, tertiary lymphoid structures [TLS ], and macrophages), 2) signatures of responses to immune checkpoint therapy, 3) interferon (IFN) signaling, and 4) Hoshida’s S1 class. Immunophenotyping of tumor sections also showed high PD-1 and PD-L1 protein expression and significantly enriched presence of TLS in the “immune class” (Fig. 3).
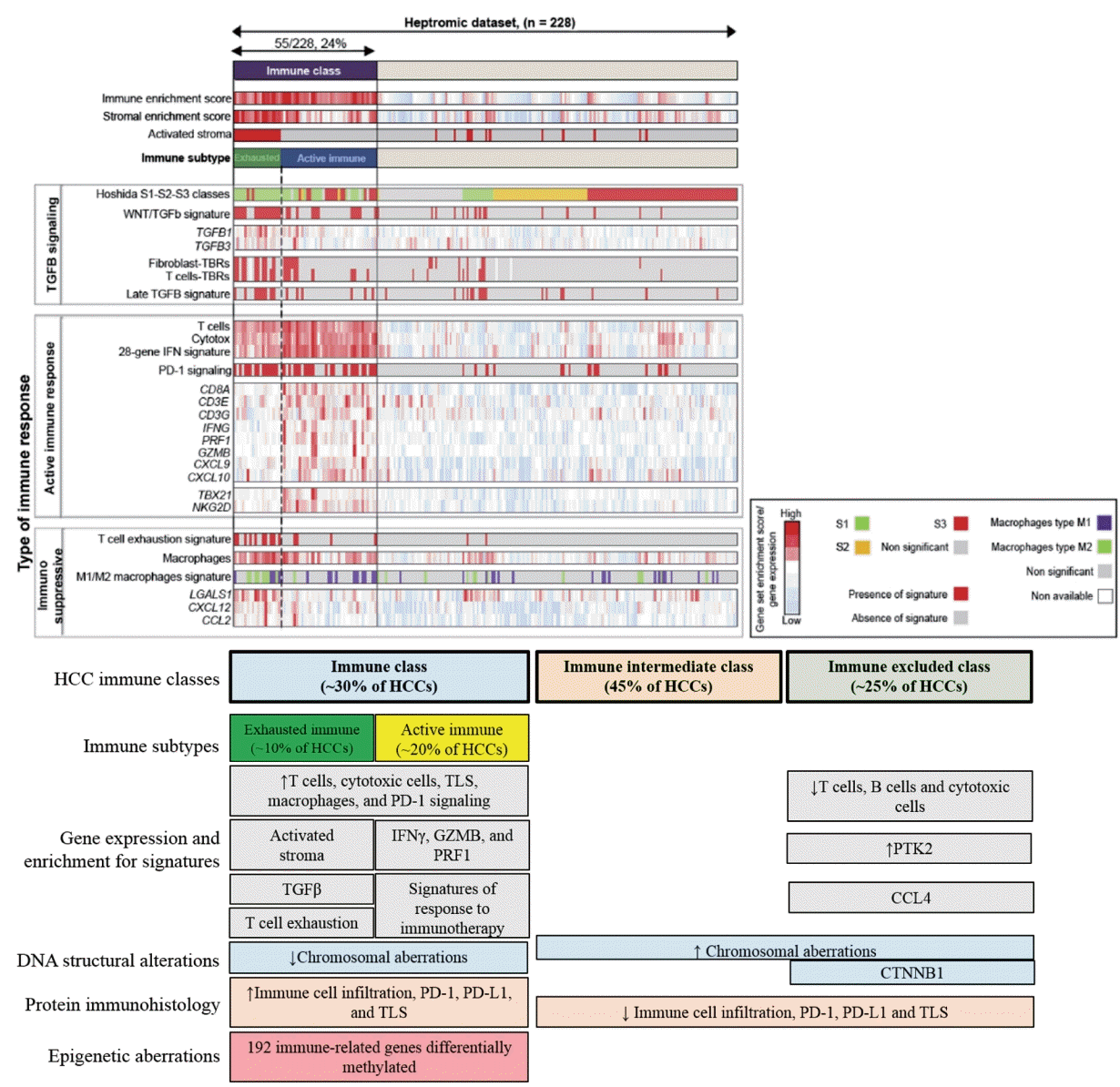 | Figure 3.Classification of hepatocellular carcinomas based on immune classes. Immune classes are characterized by high immune tumor infiltration levels and are classified into two subgroups. The “active immune” subgroup has markers for adaptive T-cell responses and has molecular traits responsive to immunotherapies, while the “exhausted immune group” has stromal and TGF-β activation. HCC, hepatocellular carcinoma; CCL4, C-C motif chemokine ligand 4; CTNNB1, catenin beta 1; IFN-γ, interferon-gamma; GZMB, granzyme B; PD-1, programmed cell death 1; PD-L1, program cell death 1 ligand 1; PRF, perforin 1; PTK2, protein tyrosine kinase 2; TGF-β, transforming growth factor-beta 1; TLS, tertiary lymphoid structures. Adapted from reference [60] and reference [62] with permission.
|
The infiltration of immune cells in tumors has a positive or negative effect on patient prognosis depending on tumor stroma modification [61]. Immune cells can destroy the homeostasis between cell proliferation and cell death through interactions with the extracellular matrix and fibroblasts, leading to epithelial-mesenchymal transition, invasion, and metastasis. The “immune class” was sub-classified into two classes. The first class, called the “active immune response” subtype, was observed in 67% of the “immune class”, and was characterized by a lack of activated stroma. The significant enrichment of T-cells and active interferon signatures, including adaptive immune response genes such as granzyme B (GZMB ), IFN -γ, CD8A , and IFN signatures predictive of pembrolizumab response, were the hallmarks of this subtype. The presence of activated stroma was observed in the “exhausted immune response” subtype.
Conversely, this subtype was characterized by tumor-promoting signals such as the T-cell exhaustion signature, defined by TGF -β-regulated network activity and M2 macrophages. TGF -β regulates tumor-stroma interaction, angiogenesis, and metastasis. Immunosuppressive factors such as galectin (LGALS1 ), C-X-C motif chemokine ligand 12 (CXCL12 ), and C-C motif chemokine ligand 2 (CCL2 ) were also observed in this subtype while essential NK cell activators such as GZMB , IFN -γ, T-Box 21 (TBX21 ), and NKG2D were significantly downregulated. The tumor recurrence rate was higher in the “exhausted immune response” group than in the “active immune response” group. Although this study is not directly applicable to clinics, immune classifiers could help identify patients who might respond better to immunotherapies and those who need additional molecular inhibitors such as the TGF -β inhibitor.
In another study, the authors analyzed the immune microenvironment of 919 regions from 158 HCCs. This study showed that in addition to the currently known histopathological and molecular subtypes, the classification of immune microenvironments into immune-high, immune-mid, and immune-low had a prognostic impact depending on the subtypes [62]. Intratumor heterogeneity among the immune subtypes was observed in 50% of cases, reflecting the multistep nature of hepato-carcinogenesis. A better prognosis was observed only in the “immune-high” subtype within poorly differentiated, high-grade HCC patients with positive CK19K , SALL4 , or both. In contrast, immune subtypes did not have prognostic impacts in well- to moderately differentiated HCCs and WNT/β-catenin HCC. The “immune-high” subtype is characterized by the co-infiltration of T and B plasma cells and showed an association with Hoshida’s S1/Boyalt’s G2 molecular subclass, PD1 positivity in CD8 + T-cells was significantly associated with PD-L1 expression in the macrophages. Progression from well-differentiated to moderately differentiated HCCs was associated with a decreasing CD8/CD3 ratio and CD56+ NK-/NKT -cell infiltration without significant changes in total T-cell infiltration. However, T-cell infiltration obviously changed during the development from moderately differentiated to poorly differentiated HCC.
2. Biomarkers associated with responses to regorafenib
The most comprehensive analysis for predicting the response to regorafenib was performed using plasma and archival tumor samples from the RESORCE trial for protein, miRNA, and genetic biomarkers [63]. Mutations in CTNNB1 were observed only in progressors, but not in responders, while VEGFA amplification based on tissue analysis was observed in responders but not in progressors. This is an important finding as VEGFA is the ligand for VEGFR , a target for regorafenib. Increased baseline plasma levels of AFP and c-MET were independent of regorafenib treatment responses, but instead five newly identified plasma proteins (ANG-1 , cystatin B , LAP TGF -β1, LOX -1 , and MIP-1α) showed significant associations with increased overall survival for regorafenib treatment. Except for ANG-1 , which is associated with angiogenesis and tumor progression, other proteins such as LAP TGF -β1 are known to be precursors to TGF -β and MIP -1α, which induce immune cell infiltration and are not direct targets of regorafenib indicating indirect communication between the biomarkers and regorafenib [64]. Levels of nine plasma miRNAs (increased levels for miR-30A, miR-122, miR-125B, miR-200A, and miR-374B; decreased levels for miR-15B, miR-107, and miR-320; absence of miR-645) showed significant associations with overall survival time with regorafenib treatment. Except for miR-122 and miR-200A which were studied extensively in HCCs, other miRNAs remain unknown and hence these findings set the stage for further prospective validation.
3. Prevention of HCC recurrence with sorafenib as adjuvant treatment
One of the most striking results, based on the BIOSTORM cohort of a subgroup of 188 patients representing 21% of the 900 STORM patients treated with surgical resection, resulted in the development of a multi-gene signature that predicted recurrence-free survival (RFS) on sorafenib [65]. Tumors from patients showing negative phospho-ERK hepatocyte staining showed increased RFS compared with those of patients in the placebo group. Phospho-ERK is associated with angiogenesis and apoptosis and could be used as a marker for recurrence with sorafenib treatment. In addition, the absence of nuclear phospho-VEGFR2 tumor staining presented a non-significant trend for better RFS with sorafenib.
A novel 146-gene expression signature composed of 87 poor-prognosis genes and 59 good-prognosis genes could precisely discriminate patients benefiting from sorafenib. This signature classified 30% of cases as “Sorafenib RFS responders”, and this group showed downregulation of poorprognosis pathways, such as KRAS , deregulation of bile acid/lipid metabolism-related signaling, and immune-related processes. In this group, the immune profile was enriched with signatures capturing the presence of B cells, CD4 + T-cells, and its derivatives (TH1, TH2, and follicular helper T-cells). “Immune class” [60] associated features (CD8 +, effector memory, and central memory T-cells and tumor-associated TLS ) were excluded but rather the innate markers, such as activated mast cells and cytolytic NK cells (NKCD56dim ), were enriched in “Sorafenib RFS responders” while activated macrophages and components of major histocompatibility complex were absent. Furthermore, this group was negatively correlated with the IFN gene signature, which predicts responses to immunotherapies in other cancers.
“Nonresponders” were characterized by the activation of signaling pathways with poor outcomes (PI3K-AKT-mTOR , KRAS , MAPK , IGF1R , and Notch ), higher mRNA AFP levels, microvascular invasion, and HCV-related HCC. For immune profiles, the “nonresponders” captured the “immune class” [60] traits characterized by the CD8 + T-cells, TLS , and PD1 signaling along with the “immune exclusion” subtype (i.e., CTNNB1 class). Unlike other cancers such as breast and colorectal cancers, where more than 20 predictive biomarkers have entered clinical practice, sorafenib and other targeted therapies are not currently in use for HCCs.
Go to :
CONCLUSION
Recent advances in the area of genomic profiling have increased our understanding of hepato-carcinogenesis, leading to the discovery of the genetic as well as epigenetic alterations required for tumor development and progression. A closer step towards personalized medicine was made following the identification of several subclasses that may respond better to targeted or immunotherapy based on comprehensive genomic and proteomic analyses. However, challenges lie with genes such as TERT , TP53 , and CTNNB that are undruggable. Applying integrated, comprehensive molecular, and histopathological immune features are important to design future clinical trials to overcome this challenge, and the combination of novel therapies could be an alternative to overcome the present drawbacks in the management of HCC.
Go to :
 XML Download
XML Download