

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Post-transplantation lymphoproliferative disease (PTLD) is one of the most common neoplastic diseases in recipients of solid organ transplantation (SOT) [1,2]. Previous reports have demonstrated that approximately 70% of PTLD cases are associated with an Epstein-Barr virus (EBV) infection and the use of immunosuppressive drugs [2]. A mismatch between EBV seropositive donor and EBV seronegative recipient can result in the development of PTLD with a 4.7-fold higher hazard ratio compared to the matched EBV seropositive recipients [2]. Though the exact pathogenesis is unclear, EBV-associated PTLD may be caused by the decrease in EBV-specific cytotoxic T-cells in recipients and the proliferation of EBV-infected B cells from the donor [3]. Life-long intake of immunosuppressive agents increases the incidence of PTLD after liver transplantation. Individual medications that are reported as potential risk factors for PTLD are anti-thymocyte globulin, calcineurin inhibitors, anti-CD3, tacrolimus, and cyclosporine [2,4-7].
Approximately half of the cases of reported PTLD occur in transplanted organs, such as the heart, lung, and liver [6,8]. As the survival rates for SOT patients have increased recently, the majority of PTLD cases are late onset cases [9]. Early onset PTLD, developing within the first six months after transplantation, rarely occurs after liver transplantation [10]. Here, we report an early-onset EBV-associated polymorphic PTLD in a graft liver, which was initially suspected to be a necrotizing abscess with central necrosis by contrast-enhanced abdominal magnetic resonance imaging (MRI), but diagnosed as polymorphic PTLD after 18F-fluorodeoxyglucose positron emission tomography (18F-FDG PET) and pathologic findings.
Go to : 
CASE REPORT
1. History and presentation
The present case report was approved by the Institutional Review Boards (Seoul St. Mary’s Hospital, KC19ZESI0149). A 54-year-old male patient was admitted for the evaluation of a hepatic mass. The patient had undergone deceased-donor liver transplantation (LT) for hepatocellular carcinoma (HCC) (Modified UICC 5th stage II, T2N0M0, Barcelona Clinic Liver Cancer stage: A) due to chronic hepatitis C virus (HCV) infection. The patient had been diagnosed with HCC three years prior to liver transplantation and had undergone trans-arterial chemoembolization nine times. At the time of liver transplantation, the viable tumor was 1 cm in size. After surgery, the patient was prescribed 400 mg cyclosporin, 1,000 mg mycophenolate mofetil, and 5 mg prednisolone daily. Five months later, the patient was admitted to the hospital for a follow-up examination during a state of complete remission of HCC with no specific symptoms.
2. Investigations
Investigations revealed normal ranges of complete blood count, blood chemistry, and tumor markers and elevated serum lactate dehydrogenase levels (456 μg/dL). HCV RNA level was 5,452,120 IU/mL indicating a recurrence of HCV. Contrast-enhanced abdominal MRI detected a 7 cm irregular necrotizing mass with rim enhancement in the right hemiliver mimicking a necrotizing abscess (Fig. 1A). 18F-FDG PET showed a single hypermetabolic malignant tumor with central hypometabolism (Fig. 1B). Ultrasound-guided biopsy of the mass was performed for diagnosis. Biopsy of the mass demonstrated a polymorphic PTLD with central necrosis (Fig. 2A). The malignant cells showed a B cell phenotype with plasmocytic differentiation (CD20, CD38 and IgG positive, kappa light chain restricted) (Fig. 2B, C). Most of the malignant cells were positive for Epstein-Barr encoding region (EBER) in situ hybridization (Fig. 2D). Serum EBV DNA level was 20,368 IU/mL, which had been negative before liver transplantation. A retrospective review of the blood test results of the deceased donor revealed that the donor was seropositive for the viral capsid antigen of EBV. A bone marrow biopsy was performed for the patient, and findings were normal. The international prognostic index was calculated and found to be one point (low-risk) and Ann Arbor staging was evaluated as stage I (single lymph node region or one extra-lymphatic site involvement).
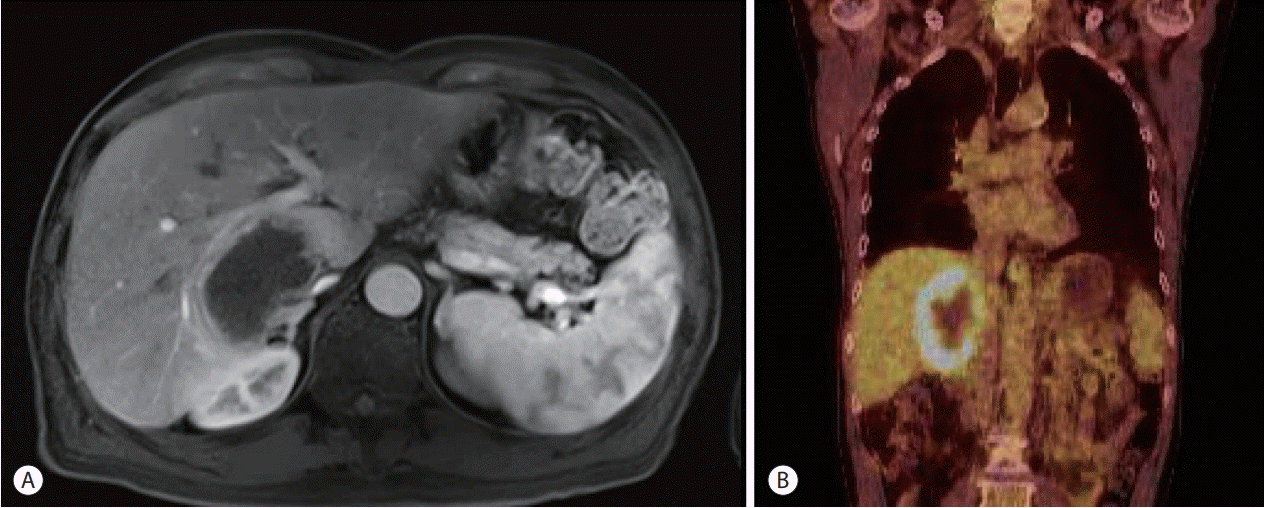 | Figure 1.Radiologic findings of the necrotizing mass in the graft liver. (A) Contrast-enhanced abdominal magnetic resonance imaging (portal phase) detected a 7 cm irregular necrotizing mass with rim enhancement in the right hemi-liver mimicking a necrotizing abscess. (B) 18F-fluorodeoxyglucose positron emission tomography shows a single hypermetabolic malignant tumor with central hypometabolism.
|
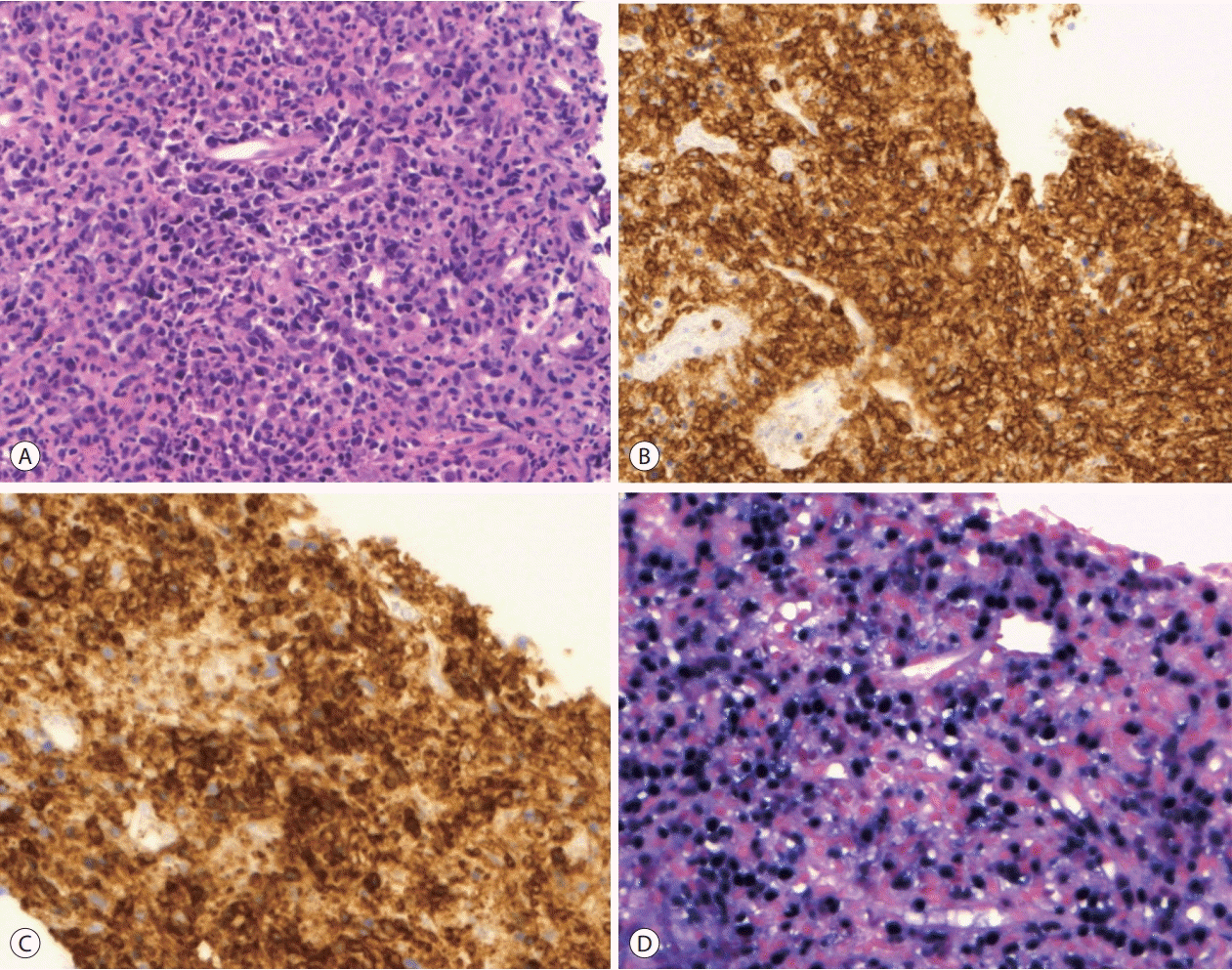 | Figure 2.Pathologic findings of the necrotizing mass in the graft liver (original magnification ×200). (A) H&E staining demonstrates a polymorphic post-transplantation lymphoproliferative disease with central necrosis. (B) Immunohistochemistry of CD20 shows positive cytoplasmic staining. (C) Immunohistochemistry of CD38 shows positive cytoplasmic staining. (D) In situ hybridization of Epstein-Barr-encoding region of RNA shows that most of the malignant cells are positive for the hybridization.
|
3. Treatment and outcome
The patient underwent direct-acting antiviral treatment (ledipasvir, sofosbuvir, and ribavirin) for recurrent HCV infection while simultaneously discontinuing cyclosporin and mycophenolate mofetil. As a substitute for immunosuppressants, 2.5 mg everolimus was applied. After completion of the anti-HCV therapy, chemotherapy was started. Four cycles of 600 mg rituximab were administered every four weeks, which resulted in a decrease in the PTLD lesion to 5 cm in contrast enhanced computed tomography (CT) imaging. Eight sequential cycles of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R-CHOP) were administered in 6 months without prednisolone because of a history of gastric ulcer bleeding. After the completion of chemotherapy, response evaluation with contrast enhanced CT imaging and 18F-FDG PET revealed a further decrease in PTLD lesion and no visible 18F-FDG uptake (Fig. 3). At this time, EBV-DNA was not detected.
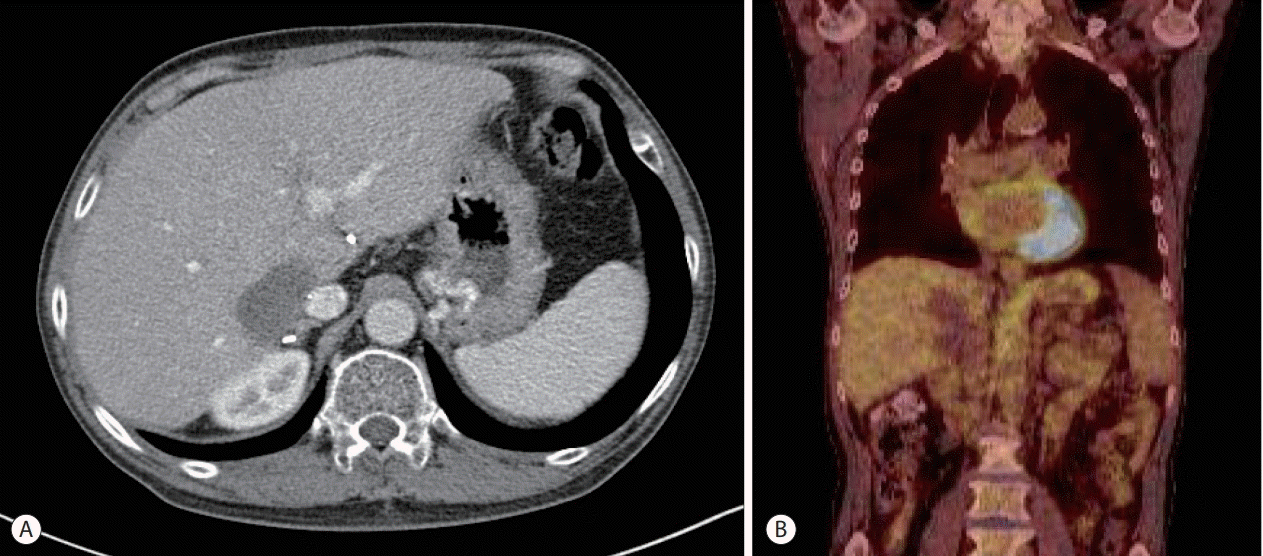 | Figure 3.Follow-up imaging findings after treatment. (A) Contrast-enhanced abdominal computed tomography imaging of the graft liver shows a dramatic decrease in the post-transplantation lymphoproliferative disease (PTLD) lesion after treatment. (B) 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography imaging reveals no visible 18F-FDG uptake in the PTLD lesion after treatment.
|
Go to :
DISCUSSION
The highest incidence of PTLD following SOT occurs in intestinal and multi-organ transplants (5-20%), and the lowest incidence occurs in kidney and liver transplants (1-5%) [6]. In comparison to other organ transplantations with prolonged use of immunosuppressants, PTLD associated with liver or kidney transplants occurs at a lower rate due to the reduced dose and administration duration of immunosuppressants [11]. Previous reports demonstrated that PTLD after liver transplantation appeared most commonly in liver grafts (approximately 20%) [12].
PTLD following liver transplantation typically occurs more than one year after the transplant in adults, because this is the period where patients receive long-term immunosuppressant treatment [10]. Generally, 60-80% of all PTLD cases are EBV-related [13], and comprehensive analysis of PTLD cases after liver transplantation demonstrated that EBV-related cases account for 80% of all PTLD cases after transplantation. Interestingly, 91% of early-onset PTLD cases were associated with EBV infection, although only 66% of late-onset PTLD cases were EBV-related [14]. Since EBV is a γ-herpesvirus that stimulates B cell proliferation and transformation, 85% of PTLD cases are B-lymphocytic in origin [14]. After successful treatment of the primary infection, EBV persists in infected B cells and establishes a latent infection. Pharmacologic immunosuppression after transplantation results in the decreased frequency and functional impairment of EBV-specific cytotoxic T-cells [12]. In this setting, EBV-driven lymphoproliferation with type 3 latency may occur and is labeled PTLD [12].
HCV infection has also been reported as a possible risk factor for non-Hodgkin’s lymphoma, especially for B cell lymphoma [15]. The possible pathophysiology of HCV infection and PTLD suggests that chronic stimulation of the immune system because of HCV infection may cause clonal B cell expansions and lymphoproliferative conditions [16]. However, findings from multiple studies indicate that the role of HCV as a risk factor for PTLD remains controversial. A retrospective cohort study for SOT patients with diagnosed PTLD during follow-up examination reported that HCV was not a major risk for PTLD in both liver and non-liver transplantations [16].
The diagnosis of PTLD following liver transplantation depends upon the findings from a liver biopsy. Classification of PTLD requires immunophenotyping for assessing light chain class restriction and lymphoid subsets, histopathology, and EBER in situ hybridization [17]. Based on the pathological findings, the World Health Organization classifies PTLD into four categories: 1) early lesions (plasmocytic hyperplasia, infectious mononucleosis-like PTLD), 2) polymorphic PTLD, 3) monomorphic PTLD (B cell neoplasms, T-cell neoplasms), and 4) classical Hodgkin’s lymphoma-type PTLD [17]. Polymorphic PTLD, as seen in the present case, involves varying differentiation stages. As presented in this patient, the infiltrate causes the underlying tissue architecture to be effaced and destructed with malignant features, such as necrosis and high mitotic rate with nuclear atypia [18]. Moreover, imaging is often helpful for the diagnosis of PTLD, and 18FFDG PET has been considered more sensitive than contrastenhanced CT and MRI [18]. In this case, contrast-enhanced MRI image findings coincided with the interpretation of the lesion as a necrotizing abscess due to low attenuation and signal at the center of the lesion with rim enhancement. However, 18F-FDG PET findings showed intense 18F-FDG uptake at the margins of the lesion, suggesting a strong possibility of PTLD with central necrosis.
Although there is no standard treatment approved for the management of PTLD due to its clinical and pathological heterogeneity, the initial treatment typically involves a reduction in immunosuppression and a renewal of immune surveillance. The restoration of immune function allows the body to control EBV-infected proliferating cells [6]. Both European and American guidelines also suggest a reduction in anti-proliferative agents such as mycophenolate mofetil and azathioprine, and switching to a mammalian target of rapamycin inhibitor, such as everolimus in this case [3]. In the cases of CD20-positive PTLD with extrahepatic invasion or resistance to immunosuppressant reduction, rituximab is considered as the first-line treatment [19]. The treatment response of first-line rituximab appeared to be 40-65% [19]. Early introduction of rituximab in patients with PTLD is reported to confer better survival outcomes [20]. In cases of rituximabresistant PTLD or high-grade lymphoma or multi-organ involved PTLD, R-CHOP is recommended [6]. For CD20-positive PTLD patients with rituximab-included regimen resistance, cytotoxic chemotherapy is considered a possible modality; however, the overall prognosis appeared to be worse when compared to that with rituximab-responsive PTLD [4]. Other treatments such as surgical resection and radiotherapy for local control may be recommended for patients with resistance to the first-line treatment, but they may carry adverse side effects [4].
Prognosis of PTLD following SOT is poor with a 60% and 40% 3-year and 5-year overall survival rate, respectively [20]. Hence, PTLD in SOT recipients should be promptly suspected based on clinical presentations and EBV serological state. However, it is difficult to reliably diagnose early-onset PTLD and initiate treatment due to the heterogeneity of clinical presentations. In conclusion, polymorphic PTLD in liver transplant recipients, less than one year after the transplantation, is rare but can cause fatal complications. Patients should undergo regular follow-up examinations for EBV serologic status. Atypical radiologic presentations, as in this case, should not be overlooked and should be carefully investigated for the possible occurrence of early-onset PTLD.
Go to :
 XML Download
XML Download