

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related deaths, with increasing incidences worldwide [1]. Although several attempts have been made to develop new drugs for HCC, these have been largely unsuccessful. Recently, numerous phase III clinical trials investigating sunitinib, brivanib, erlotinib, or cixutumumab have failed [2-6]. Only lenvatinib showed non-inferior overall survival (OS) than sorafenib [7]; treatment with regorafenib, an oral multi-kinase inhibitor targeting angiogenic, stromal, and oncogenic receptor tyrosine kinases, resulted in improved OS in sorafenib-treated patients showing cancer progression [8]. However, these small molecular inhibitors have limited use, as only 30% of patients have the mutations that these therapeutics target [9].
HCC is an inflammation-induced cancer which represents a unique target for immune-based approaches [10]. Immune-based approaches focusing on vaccination, treatment with cytokines, or non-specific T cell activation in HCC have mostly been unsuccessful [11,12]. However, United States Food and Drag Administration (FDA) approval of immune checkpoint inhibitors brought about a dramatic change in the era of immuno-oncology. Nivolumab, which blocks programmed cell death protein-1 (PD-1) pathway, has been approved as a second-line therapeutic agent for advanced HCC [13], and studies to evaluate its use as a first-line treatment for advanced HCC are ongoing.
Herein, we describe the mechanisms of immune interventions used for the treatment of HCC. We summarize data from recent studies, ongoing clinical trials, and adverse events resulting from immunotherapeutic use. In addition, we discuss the development of immunotherapeutics that may be alternative treatment options for HCC in the future.
Go to : 
MECHANISMS OF IMMUNE TOLERANCE AND SUPPRESSION OF HCC
The liver has a specific blood supply containing 25% hepatic arterial flow and 75% portal venous flow. Portal venous flow drains into the sinusoids with very low vascular resistance and is loaded with nutrients and microbial antigens from the intestines. Therefore, many microbial antigens are in contact with non-parenchymal cells and immune cells, including hepatic stellate cells, liver sinusoidal endothelial cells, dendritic cells (DCs), Kupffer cells, and lymphocytes. To prevent hyperstimulation caused by intestinal antigens, the liver has several self-tolerance mechanisms: 1) decreased expression of costimulatory immune receptors such as B7-1 and B7-2; 2) upregulation of PD-1 and cytotoxic T lymphocyte antigen-4 (CTLA-4) immune check point inhibitors on hepatic antigen presenting cells (APCs) [14]; and 3) secretion of the cytokines, such as interleukin (IL)-10 and transforming growth factor-β [15,16]. Infection with hepatitis B and C viruses leads to frequent chronic inflammation in the liver, resulting in dysregulation of T cell responses [17]. Moreover, tumor growth favors this dysregulation, which is already present in HCC patients [18].
The ability of tumor cells to evade the immune system is a key for transformation of non-tumor cells into malignant cells [19]. The immune cells and tumor cells interact through a dynamic process known as immunoediting, which has three phases: 1) the elimination phase, in which transformed cells are destroyed by immune cells, such as cytotoxic T lymphocytes, and natural killer (NK) cells; however cancer cells change their phenotypes making them resilient to elimination; 2) the equilibrium phase, in which cancer cells begin to reduce their immunogenicity; and 3) the escape phase, in which cancer cells fully escape immune-mediated killing mechanisms and eventually form tumors. Tumor cells can overexpress immune checkpoint molecules, which bind to their receptors on T cells and inhibit T cell activation.
Three main strategies have been used to improve the tumor-specific immune response: 1) adoptive immunotherapy, in which immune cells recognizing HCC antigens are infused into the patient; 2) indirect immunological strategies including treatment with cytokines and monoclonal antibodies (mAbs) against immune checkpoint proteins and cancer vaccines that activate the immune system; and 3) indirect nonimmunological strategies, including oncolytic viruses, antigen-encoding mRNA, and metronomic chemotherapy (Fig. 1).
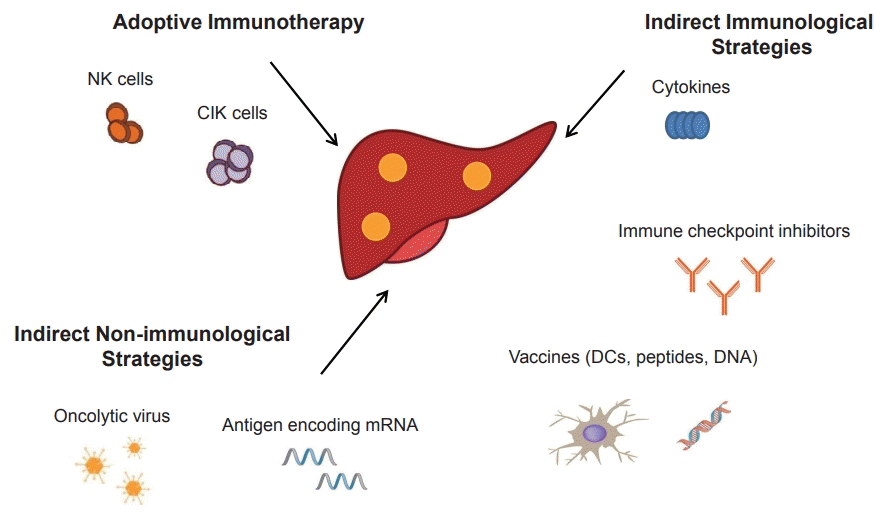
Go to :
ADOPTIVE IMMUNOTHERAPY
Adoptive cell therapy kills patient’s own lymphocytes, including cytokine-induced killer (CIK) cells, tumor-infiltrating lymphocytes (TILs), NK cells, and chimeric antigen receptor (CAR) T cells to eliminate cancer cells. In this treatment, autologous lymphocytes are stimulated with cytokines or tumor antigens and cultivated ex vivo before being reinfused back into a patient [20].
1. CIK cells
CIK cells are heterogeneous non-major histocompatibility complex (MHC)-restricted cytotoxic T cells mainly comprising CD3+CD56+, CD3−CD56+, and CD3−CD56+ cells [21]. CIK cells are expanded ex vivo from peripheral mononuclear cells stimulated with anti-CD3 antibodies, IL-2, and interferon gamma. These cells have many characteristics of terminally differentiated CD8+ effector memory cells, and simultaneously recognize HCC cells in an MHC class Ι-restricted manner. In a phase III study using adjuvant immunotherapy with activated CIK cells after radical resection for HCC, recurrence-free lifespan and OS were prolonged [22]. In an extended 5-year follow-up study, patients who received an adjuvant CIK cell immunotherapy for HCC showed significant improvement in recurrence-free and OS that lasted for over 5 years without additional booster treatments [23]. A meta-analysis of eight randomized clinical trials and six prospective studies revealed that CIK treatment increased the rate of patient survival, but did not prolong progression-free survival [24].
2. TILs
TILs are isolated from tumor tissues and are cultured and activated using anti-CD3 antibodies and IL-2 ex vivo . TILs are considered to have more specific anti-tumor immunological activities than non-infiltrating lymphocytes [25]. A phase I study was performed to determine the safety and efficacy of TILs in patients with HCC. The autologous TILs had relatively less toxicity, suggesting that TILs can be used safely as a treatment for HCC [26]. TILs have not yet been well characterized, mainly due to difficulties in purifying and expanding them.
3. NK cells
Human NK cells (CD56+CD3-) are major players in innate immunity and are involved in defense mechanisms against cancer cells [27]. NK cells kill tumor cells directly without prior sensitization or MHC restriction. However, they lack the ability to target cancer cells and can harm normal hepatocytes. A clinical trial (NCT02008929) has been performed to examine the efficacy of adoptive NK cell transfer in preventing HCC recurrence after curative therapy, but the results are not yet available. Another phase I clinical trial (NCT01147380) examined the feasibility and safety of adoptive transfer of IL-2-activated NK cells obtained from cadaveric donor liver grafts to liver transplant recipients with HCC. No severe adverse effects were observed in the 18 patients who received the liver NK cells. A phase II clinical trial (NCT02725996) to determine the efficacy and safety of autologous NK cells in patients who underwent curative resection for HCC is ongoing.
4. CAR T cells
CAR T cells are genetically modified T lymphocytes that specifically target tumor-associated antigens and kill cancer cells in an MHC-independent manner [28]. CAR T cells consist of three major components: the extracellular antigen-binding domain, the intracellular signaling domain, and the hinge area [29]. The antigen-binding domain is a single chain fragment variable (scFv) region, which contains a heavy (VH) and a light (VL) connected by a linker fragment chain, derived from mAbs (Fig. 2). The signaling domain has immunoreceptor tyrosine-based activation motifs (ITAMs), such as CD3ζ and FcεRIγ. The antigen-binding and signaling domains are connected by a hinge area. The CAR gene is generally transduced into T cells using a lentiviral vector. The second- and third-generation CARs, in which costimulatory molecules (CMs) are added, generate highly proliferative, persistent, and potent cytotoxic T cells. A protein, glypican-3 (GPC3) is expressed abundantly in HCC cells, and frequently used as a target for CAR T cells. Two phase I clinical trials (NCT02395250 and NCT02723942) have been conducted to determine the safety of anti-GPC3 CAR T cells transfer into HCC patients; however, the results are not yet available. A phase I clinical trial (NCT03672305) to evaluate the efficacy and safety of transferring c-Met/programmed cell death ligand 1 (PD-L1) CAR T cells in patients with HCC is ongoing.
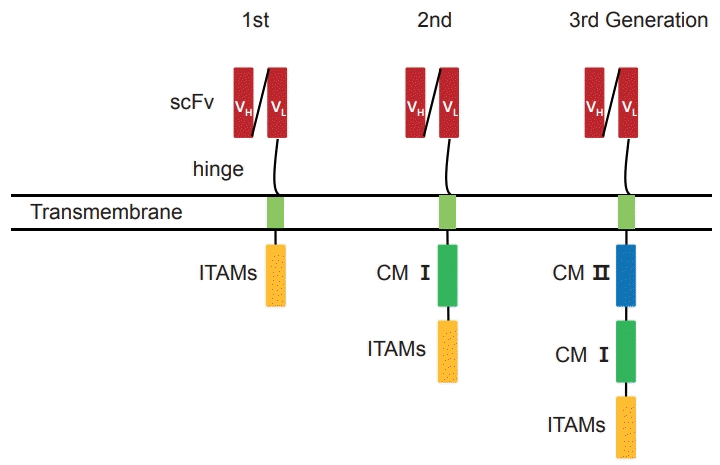
Go to :
INDIRECT IMMUNOLOGICAL STRATEGIES
Immune checkpoint proteins include CTLA-4, PD-1, PD-L1, T cell immunoglobulin and mucin domain 3 (TIM-3), and lymphocyte activation gene 3 (LAG-3) (Fig. 3) [30]. Among these, inhibitors of CTLA-4 and PD-1 have been well-characterized in HCC (Table 1).
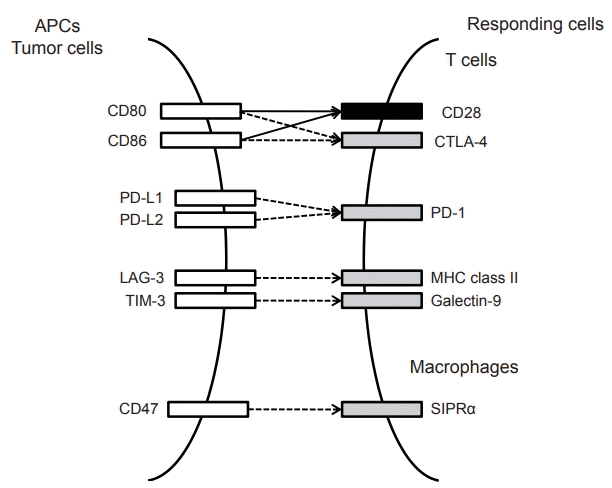 | Figure 3.Interaction between major immune co-stimulatory and inhibitory molecules and their cognate receptors. Co-stimulatory and co-inhibitory molecules are indicated by closed and open boxes, respectively. Costimulatory and inhibitory signals are indicated by filled and hatched arrows, respectively. APC, antigen presenting cell; PD, programmed cell death; CTLA, cytotoxic T lymphocyte antigen; LAG, lymphocyte activation gene; TIM, T cell immunoglobulin and mucin domain; MHC, major histocompatibility complex; SIPR = sphingosine-1-phosphate receptor.
|
Table 1.
Recent clinical trials of immune checkpoint inhibitors in HCC
![]()
Cancer vaccines induce tumor-specific immune responses by inducing effector T lymphocytes that can reduce tumor burden and prevent tumor recurrence. The agents used for HCC vaccines include HCC cells, antigens, DCs, and DNA.
1. Immune Checkpoint Inhibitors
1) CTLA-4 inhibitors
CTLA-4 is predominantly expressed on activated T lymphocytes and has a high affinity for the CMs, CD80 and CD86. CD80 and CD86 are expressed on APCs and can bind to the CM, CD28. A phase II study demonstrated that the anti-CTLA-4 mAb, tremelimumab, is a safe anti-tumor and anti-viral therapeutic agent for hepatitis C virus-induced HCC, with a partial response (PR) rate of 17.6%, a disease control rate of 76.4%, and time-to-progression (TTP) of 6.48 months [31]. In another clinical trial that included patients with advanced HCC, a combination therapy of tremelimumab and radiofrequency ablation (RFA) increased the number of intra-tumoral CD8+ T cells and reduced HCV viral loads [32]. A phase I trial (NCT01853618) to determine the safety and effectiveness of tremelimumab with chemoembolization or RFA is ongoing.
2) PD-1 inhibitors
PD-1 is expressed in T cells, B cells, NK cells, mononuclear cells, and DCs. PD-1 inhibitors block the binding of PD-L1 and PD-L2 to PD-1, preventing the suppression of T cell activity. A phase I/II study has demonstrated the safety and efficacy of nivolumab, a fully humanized IgG4 monoclonal antibody to PD-1, in patients with advanced HCC with 5% complete response (CR) and 18% PR [33]. Moreover, response durations for CR, PR, and stable disease were 14-17+ months, <1-8+ months, and 1.5-17+ months, respectively; the OS rate at 6 months was 72%, implicating that nivolumab helped sustain tumor-specific immune responses. No serious hepatic dysfunction or autoimmune disease occurred. A phase III trial (NCT02576509) comparing nivolumab to sorafenib as a first-line of treatment in patients with advanced HCC is ongoing. Several clinical trials on nivolumab as a combination therapy are ongoing, including a phase II trial (NCT03033446) on Y90-radioembolization with nivolumab in Asians, a phase I trial (NCT03299946) with neoadjuvant nivolumab + cabozantinib (multiple tyrosine kinase inhibitor), and a phase II trial (NCT03510871) on neoadjuvant nivolumab + ipilimumab (anti-CTLA-4 mAb).
A phase III trial (NCT02702401), on pembrolizumab (MK-3475, Merck & Co., Inc., Rockville, MD, USA) was conducted in patients with advanced HCC who had been systemically treated previously and a subsequent phase III trial (NCT03062358) is planned for Asian patients. In Japan, a phase III global trial (NCT03412773) was designed to compare the efficacy and safety of tislelizumab (BGB-A317) vs. sorafenib as a first line systemic treatment in patients with unresectable HCC.
On 9 November 2018, FDA accelerated approval to pembrolizumab (KEYTRUDA, Merck & Co.) for patients with HCC who had been previously treated with sorafenib. The approval was based on KEYNOTE 224 (NCT02702414), a single-arm, multicenter phase II trial enrolling 104 patients with HCC (overall response rate of 17%). Patients were required to have either on-going disease progression, or should have completed sorafenib treatment or were intolerant to sorafenib, or have a measurable disease, or Child-Pugh class A hepatic impairment. Patients received 200 mg of pembrolizumab as an intravenous infusion every three weeks until disease progression, unacceptable toxicity or up to 24 months in patients without disease progression. The recommended pembrolizumab dose for HCC is 200 mg administered as an intravenous infusion over 30 minutes every 3 weeks. The accelerated approval of pembrolizumab in HCC is contingent on the results of a confirmatory phase III trial (NCT02702401).
3) PD-L1 inhibitors
HCC cells can evade immune surveillance by overexpressing PD-L1. PD-L1 expression is positively correlated with hepatitis B virus infection and the HCC stage [34]. Moreover, higher expressions of PD-L1 and PD-L2 in HCC tissues have been associated with poorer prognosis [35]. A phase III trial (HIMALAYA trial, NCT03298451) to assess the efficacy and safety of tremelimumab±durvalumab (mAb blocking the interaction of PD-L1 with PD-1 and CD80) as a first-line treatment in patients with unresectable HCC is ongoing.
4) Biomarker for immune checkpoint inhibitors
To find the most suited patient population for immunotherapy, it is crucial to identify biomarkers to predict the response to checkpoint blockades. The overexpression of PD-L1 is an important and widely-explored predictive biomarker for the response to PD-1/PD-L1 antibodies [36,37]. Recently, a randomized phase II trial showed that a combined positive score (CPS) of PD-L1 expressions in both tumor cells and immune cells was associated with a response to pembrolizumab [38]. The expression of PD-L1 can be considered as a dynamic process during the recognition of effective T-cell antigens. However, an evaluation at any a single time-point may not reflect an evolving immune response or predict the response to PD-1/PD-L1 pathway blockades due to the dynamic changes of PD-L1 expression. Many ongoing clinical trials are investigating the clinical usefulness of CPS for PD-L1 in predicting responses to anti-PD-1 therapy.
Many studies are exploring ideal candidates in search of biomarkers. Tumor infiltrating immune cells or molecules in the tumor microenvironment, along with PD-L1, may be important in predicting the efficacy of immune checkpoint inhibitors [39]. In addition, mismatch-repair status has also been suggested as a predictor of immune checkpoint inhibitor response [40].
5) Combined locoregional therapy and immunotherapy
Many investigators have initiated clinical trials evaluating the efficacy and safety of a combination of immune checkpoint inhibitors with locoregional therapies. RFA destroys HCC tissue leading to local necrosis. Marked inflammatory response with a dense tumor-specific T-cell infiltrate is reported to occur in such patients [41]. Mizukoshi et al. [42] reported that TAA‐specific T cells increase in 62.3% of patients after RFA, contributing to progression‐free survival (PFS). Recently, Duffy et al. [32] performed a pilot study for the safety and feasibility of tremelimumab in combination with RFA or chemoablation. PFS rates were 57.1% at 6 months and 33.1% at 12 months in 32 patients with advanced HCC (Barcelona Clinic Liver Cancer stage B, 7 patients; stage C, 21 patients, respectively). The median TTP and OS were 7.4 and 12.3 months, respectively. Minor toxicities such as pruritis were noted. Further clinical trials are ongoing to confirm the improved efficacy of locoregional therapy and immune checkpoint inhibitors (tremelimumab, NCT01853618; pembrolizumab, NCT03753659; durvalumab, NCT02821754).
2. HCC Vaccines
1) Antigen peptide vaccine
Peptide-based, tumor-associated antigens (TAAs) including alpha-fetoprotein (AFP), GPC3, SSX-2, NY-ESO-1, human telomerase reverse transcriptase (hTERT), HCA587, and melanoma antigen gene (MAGE) have been used as targets for HCC vaccines [43]. AFP and GPC3, in particular, are well-known TAAs which have been used as antigens for HCC vaccines. One phase I trial reported that use of an AFP vaccine increased T-cell-specific activity in patients with HCC [44]. In a phase II trial of an adjuvant GPC3 vaccine, patients received 10 vaccinations during the course of a year after surgery. The recurrence rate in patients who were vaccinated was significantly lower than that of the control patients (24% vs. 48%) after a one-year follow-up [45].
2) DC-based vaccine
DCs are powerful APCs that play a key role in both innate and adaptive immunity. DCs can also activate NK cells as well as T cells [46]. However, DC-induced immunity is frequently suppressed in tumor tissues, due to a low number of DCs at tumor sites, low antigen-presenting capacity of these DCs, and poor access of DCs to tumor antigens [47]. These problems can be overcome by administering ex vivo expanded DCs from peripheral blood mononuclear cells; pulsing DCs with tumor lysates, TAAs, or TAA-derived peptides; transfection of DNA constructs encoding TAAs; and fusion of DCs with tumor cells.
A phase I/IIa study, using TAA (AFP, GPC3, and MAGE)-pulsed DCs for HCC patients after primary treatment, reported that DC vaccination was an effective adjuvant treatment [48]. The same group conducted a randomized phase II trial on 156 HCC patients who were treated for HCC with no evidence of residual tumors [49]. Adjuvant DC vaccination reduced the risk of tumor recurrence in HCC patients who underwent standard treatments other than RFA. Trials using DC vaccination have shown several promising results with few adverse events, but further phase III trials are needed.
Go to :
INDIRECT NON-IMMUNOLOGICAL STRATEGIES
1. Oncolytic viruses
Oncolytic viruses are wild-type or engineered viruses which selectively replicate in tumor cells leading to tumor lysis without harming non-tumor tissues [50]. Oncolytic viruses are able to kill cancer cells directly by expanding inside cancer cells, thereby causing cell lysis. Wild-type viruses including retroviruses, varicella viruses, and Sindbis viruses can specifically infect tumors [51]. Moreover, genes that are crucial for non-tumor cells but have no functions in tumor cells can be deleted by engineering [52]. In addition, viral transcription can be limited in cancer cells by applying tumor-specific promoters [53]. After modifications by TAA-specific receptors, oncolytic viruses can effectively target tumor cells. For example, oncolytic vaccinia virus engineered with antiangiogenic genes specifically inhibit tumor angiogenesis [54].
A phase II trial tested the feasibility of two doses of JX-594 (Pexa-Vec), an oncolytic and immunotherapeutic vaccinia virus, in 30 patients with HCC. The treatment was well tolerated, and a significantly prolonged OS in the high-dose arm was seen than in the low-dose arm (14.1 months and 6.7 months, respectively) [55]. A phase III trial (PHOCUS trial, NCT02562755) comparing vaccinia virus-based immunotherapy + sorafenib vs. sorafenib alone in patients with advanced HCC is ongoing.
Go to :
ADVERSE EVENTS AFTER IMMUNOTHERAPEUTIC TREATMENT
Increasing immune system activation and blocking immune checkpoints may result in inflammatory side-effects. Immunotherapy-related adverse events commonly involve the gastrointestinal tract, endocrine glands, skin, and liver (Fig. 4) [56]. Such adverse events in patients undergoing immunotherapeutic treatment usually start within the first few weeks to months after immune checkpoint blockade treatment but can occur anytime, even after discontinuation of the treatment. Dermatologic adverse events are usually the first ones to appear. Most of the toxic effects are reversible except for the effects on the endocrine system, which may be permanent. Fortunately, deaths from such immunotherapyrelated adverse events are exceptionally rare, but deaths due to myocarditis, pneumonitis, colitis, neurologic events, and other events can occur. However, such types of adverse events may occur in different systems of the body. Therefore, multidisciplinary collaborative management of patients undergoing immunotherapy is required by healthcare providers across the clinical spectrum [57].
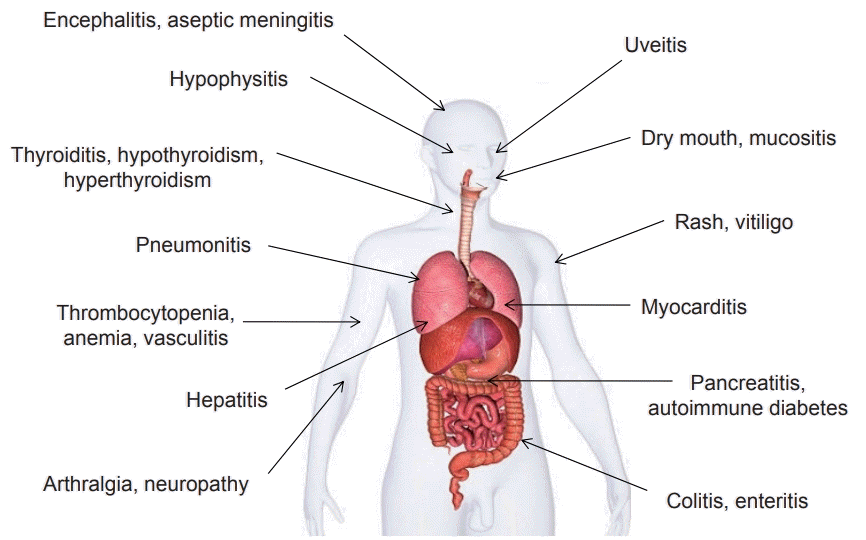
To date, no prospective trials have defined strategies for effectively managing specific immunotherapy-related adverse events. Immunosuppression therapy is used to reduce the excessive state of inflammation associated with such adverse events. Glucocorticoids are usually the first-line immunosuppressive agents used. If glucocorticoids are not effective, additional immunosuppressive agents can be used [58]. There is little data related to the safety of restarting immune checkpoint blockade treatment after a major adverse event. Retrospective studies have shown that immunotherapy-related adverse events associated with one class of agents may not necessarily reoccur during a subsequent treatment with another agent [59].
Go to :
FUTURE PERSPECTIVES IN HCC TREATMENT
Although novel molecular approaches for the treatment of HCC have been studied, only a limited improvement in survival rates has been observed. Consequently, there is an urgent need to develop new therapeutic strategies and to determine which patients would benefit from them. Immunotherapy has emerged as an alternative treatment for various malignancies. Numerous studies have shown that immunotherapy has a significant antitumor efficacy. Future studies are necessary in order to identify more specific immune targets, the most suited patient population for immunotherapy, and the best conventional therapies to be used in conjunction with immunotherapy. On November 2018, FDA accelerated approval to pembrolizumab for patients with HCC who had been previously treated with sorafenib. A confirmatory phase III trial (NCT02702401) is ongoing that might accelerate the usage of immunotherapy in HCC. Taken together, the promising therapeutic approaches mentioned in this review will see the beginning of a new era in the treatment of HCC.
Go to :
 XML Download
XML Download