

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Pharmacokinetics (PK) and pharmacodynamics (PD) are the two main areas of integrative pharmacology. PK evaluates ‘what the body does to the drug’ (absorption, metabolism, distribution and excretion of drugs), and PD studies ‘what the drug does to the body’ (biological effects of drugs over time, the relationship between drug exposure and effects, mechanism of action of drugs). The primary purpose of PK and PD modeling is to discover the biophysiological key properties of the drug (affinity, efficacy, potency and specific systemic factor), and to predict the time course and extent of drug effect under normal physiological condition, as well as specific pathological conditions [1,2]. Usually, PK studies are first performed in various in vitro and in vivo environments, and then the concentration-effect/dose-effect relationship is inferred. To identify a meaningful PK-PD relationship, it is important to fully correlate the pharmacological effect with the drug concentration of the appropriate compartment in vivo [3–5]. In this process, if hysteresis (a time lag between the measured concentration and the observed effect) is demonstrated, several PK-PD modeling approaches, such as effect compartment link model, turnover model/indirect response model, and tolerance/ rebound model, can be employed to describe it [3,6,7]. This paper introduces the semi-compartmental model, one method to collapse the hysteresis in the PK-PD relationship.
PHARMACOKINETICS
The PK model represents the time-concentration course of a drug at the measured site (usually in plasma or whole blood), or in additional hypothetical compartments which cannot be measured directly [3,4]. Compartmental PK models, that simplify complex organs and tissues into compartments and connecting lines, are most widely used [8,9]. Fig. 1 shows a three-compartment model.
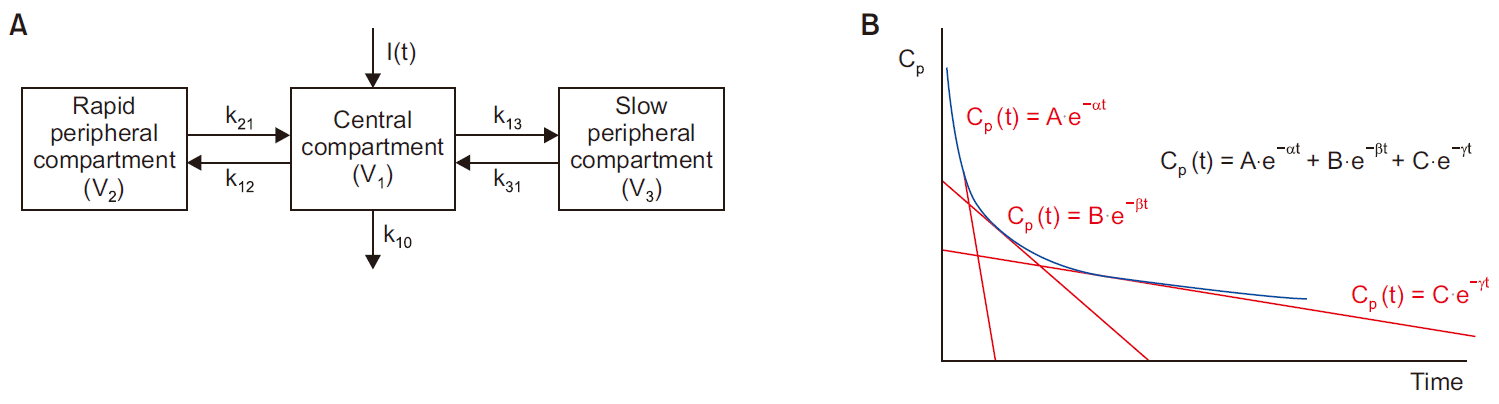
Fig. 1
Three-compartment model. (A) Scheme for three-compartment model. (B) Time-concentration curve plotted on semi-logarithmic scale in the three-compartment model following an intravenous bolus administration. I(t): drug dose (input), Vi: distribution volume of compartment i, kij: inter-compartmental fractional rate constant (micro-rate constant from compartment i to compartment j), Cp: drug concentration in plasma, A, B, C: coefficient (macro-constant), α, β, γ: exponent (macro-constant slope).
PHARMACODYNAMICS
The PD model relates the drug concentration provided by the PK model to the observed drug effect [2,3,5,7]. The most commonly used PD model for describing the non-linear concentration-effect relationship is the sigmoid Emax model (the Hill equation [7,10], Eq. 1):
where E is effect, C is drug concentration, Emax is the maximum effect, EC50 is the concentration of the drug producing half of Emax and n is the so-called steepness factor (n does not necessarily have a direct biological meaning, but it determines the slope of the curve. In the ordinary Emax model, n is equal to 1). This model can be used extensively in other areas; for example, in the receptor theory, EC50 reflects the potency of the drug in the system (the sensitivity of the organ or tissue to the drug) and Emax is the efficacy of the drug. If n is less than 1, the curve becomes hyperbolic, implying active metabolites or multiple receptor sites [5,7]. Fig. 2 presents the excitatory sigmoid Emax model including the baseline effect (E0), and the equation is functionally described as follows (Eq. 2):
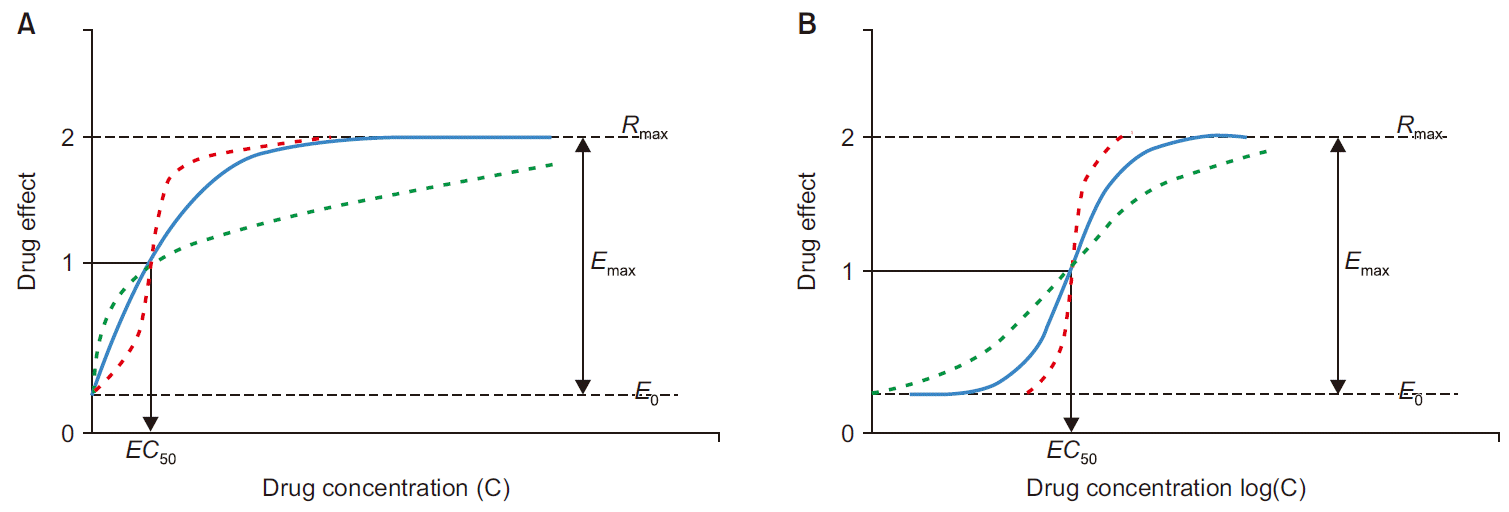
Fig. 2
The excitatory sigmoid Emax model with baseline (E0) on linear scale (A) and semi-logarithmic scale (B); when n (steepness factor) increases, the steepness of the tangent to the curve at EC50 increases (n of the red dotted line is larger than the blue solid line, and n of the green dotted line is smaller than the blue solid line). Emax: efficacy, the difference between baseline (E0) and maximum observed effect (Rmax), EC50: potency, the plasma concentration corresponding to the half of Emax. The figure is modified from the article of Gabrielsson et al. (Pharmacodynamic concepts 2016; 199-332) [7].
KINETIC-DYNAMIC (PK-PD) MODELING
Since PK and PD models share a common feature, i.e. concentration, they can be integrated to describe the overall dose-effect relationship, leading to useful insights into rational dose regimen design (Fig. 3) [3–5]. In the direct link model (Fig. 4A), the simplest form of the PK-PD model, the measured plasma concentration is directly linked to the effect-site concentration, as the equilibrium between both concentrations is assumed to be rapid and thus their ratio is constant under pharmacokinetic steady-state, as well as non-steadystate conditions. Hence, the measured plasma concentration can serve as an input function for the concentration-effect relationship, with the peak of plasma concentration and maximum effect occurring simultaneously [3,5,11].
Fig. 3
Schema of presenting of the dose-effect relationship using pharmacokinetic (PK) and pharmacodynamic (PD) models.
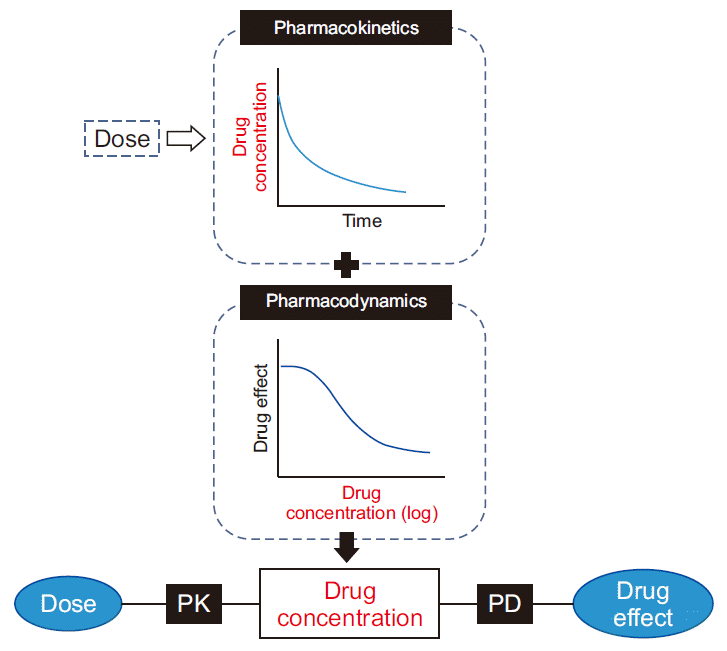
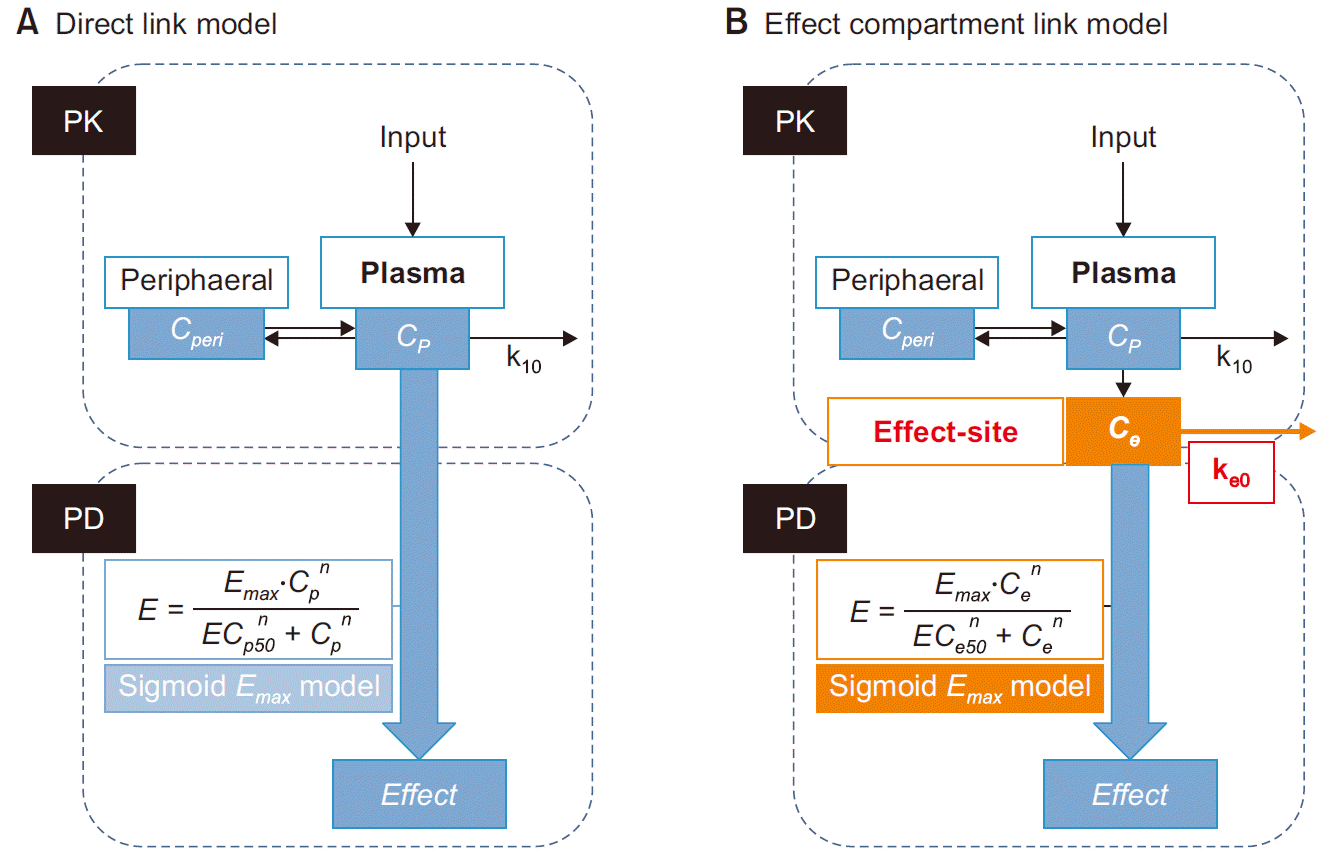
Fig. 4
Direct link model (A) vs. effect compartment link model (B). (A) In direct link model, measured plasma concentration (Cp) and observed effect (Effect) are linked directly. (B) In the effect compartment link model, an indirect link model, Cp and Effect are linked indirectly via a hypothetical effect site concentration (Ce). input: administrated drug dose, k10, ke0: first- order rate constants, Cp: measured plasma concentration, Ce: effect site concentration, Effect (E): observed effect, the effect is mediated by an arbitrarily chosen sigmoid Emax model.
However, the assumption applied to the direct link model is often difficult to employ during actual physiological situations. In case of anesthesia induction, intravenous anesthetics and neuromuscular blockers produce a peak in the blood in a short time; however, no tracheal intubation is concurrently performed since while the drug plasma concentration reaches its maximum, further time is required for the drug to reach its maximum at the effect site. As observed, when a distribution delay between the plasma concentration and the effect site concentration (hysteresis) appears, the plasma concentration fails to represent the effect site concentration (drug effect).
WHAT IS HYSTERESIS IN THE PK-PD RELATIONSHIP?
Hysteresis is a phenomenon that occur occasionally in PKPD modeling. Hysteresis can be easily comprehended by utilizing visual thinking. When the concentration-effect curve is drawn (Fig. 5), the hysteresis loop can be observed. If you draw a vertical line at the ‘concentration’ axis in Fig. 5, the line crosses the curve at 2 points, indicating two different response levels for a single drug concentration. Counter-clockwise (anti-clockwise) hysteresis implies that the observed drug effect increases over time for a given drug concentration; in clockwise hysteresis, the observed drug effect decreases over time for a given drug concentration [4,6].
Fig. 5
Concentration-effect curve with hysteresis loop. (A) Counter-clockwise hysteresis between plasma drug concentration and observed drug effect. (B) Clockwise hysteresis between plasma drug concentration and observed drug effect.
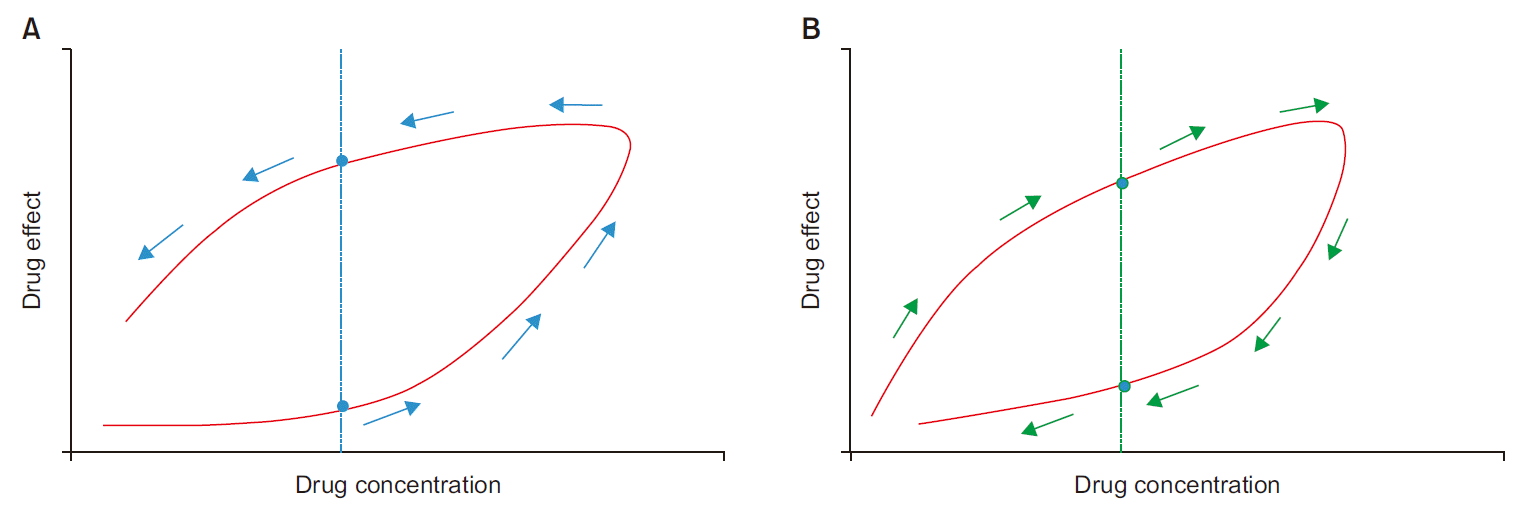
The hysteresis loop suggests that there is a time discrepancy in the relationship between the measured drug concentration and the observed drug effect. Notably, hysteresis can occur due to a consequence of different PK and PD mechanisms, including distribution delay, input-output rate change, tolerance, formation of active metabolites, timedependent protein binding, multiple receptor sites or up/down regulation of receptor, racemic drugs, and non-stereospecific assays [4,6]. To collapse hysteresis and to simplify the concentration-effect relationship, several PK-PD modeling approaches, such as effect compartment link model, turnover model/indirect response model, and tolerance/rebound model, have been utilized [3,6,7].
EFFECT COMPARTMENT LINK MODEL
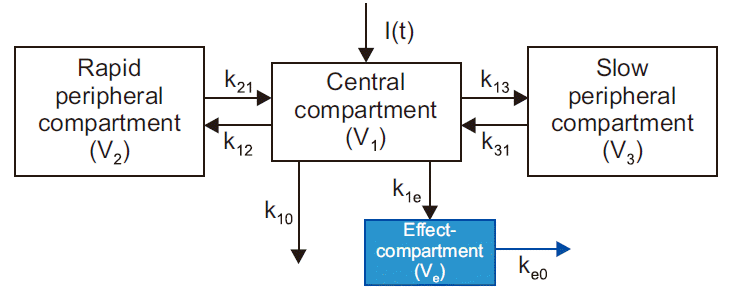
The effect compartment link model was elaborated by Holford and Sheiner [4,12,13] based on the concept of Segre [14]. This model is useful to describe the time displacement between the measured concentration and observed effect (hysteresis loop in concentration-effect curve), which often occurs due to a delayed distribution between the drug concentration in plasma and the effect site. In this model, a hypothetical effect compartment is linked to the central compartment of a PK compartment model by first-order disposition kinetics (Figs. 4B, 6). It is assumed that the effect compartment has a negligible volume compared to the central compartment, receiving a negligible amount of the drug (the amount of drug influx into the central compartment from effect compartment is also negligible). The first-order rate constant ke0, defined as the loss of drug from the effect compartment, is not directed toward any of the PK compartments, implying excretion from the body. Therefore, ke0 (called ‘equilibration rate constant’) determines the concentration equilibrium between the plasma and effect compartment (the larger ke0, the faster the equilibrium, so the effect of the drug is faster) [2,3,5–7]. The relationship between the concentration at the effect compartment (Ce) and the plasma concentration (Cp) is expressed by the differential equation as below Eq. 3:
Fig. 6
Three-compartment model with an effect-compartment. The effect compartment links to the central compartment of a threecompartment model by the first-order process. I(t): drug dose (input), Vi: distribution volume of compartment i, kij: intercompartmental fractional rate constant (micro-rate constant from compartment i to compartment j).
SEMI-COMPARTMENTAL MODEL
The semi-compartmental model is another estimation method that can be used to collapse hysteresis of the PKPD relationship, deriving the solution for the effect site concentration from the effect compartment link model. However, unlike the effect compartment link model in which PK parameters are first estimated and then PD parameters and ke0 are estimated, the semi-compartmental model does not require PK parameters (compartmental PK modeling) to estimate PD parameters and ke0 [7,15] (In ‘NONMEM’, a computer program for modeling, the effect compartment link model is named ‘sequential PK-PD modeling’ and the semicompartmental model is named ‘direct PD fit’).
There are two different solutions used in the semi-compartmental model: linear solution and log-linear solution [7,15] (In ‘NONMEM’, a linear solution is applied when the plasma concentration is increased, and the log-linear solution is applied when the plasma concentration is decreased. The control file for the semi-compartmental modeling used in ‘NONMEM’ is presented in the Appendix 1).
In the linear semi-compartmental solution, the plasma concentration (Cp) obtained by assuming a piecewise linear PK model is expressed as Eq. 5.
where
The integration of Eq. 3 defined the Cp-Ce relationship and Eq. 5 yields the solution for concentration at the effect site (Ce) given as Eq. 6:
In the log-linear semi-compartmental solution, the plasma concentration (Cp) is expressed in exponential form as Eq. 7, assuming a piecewise log-linear PK model.
where
CONCLUSION
The semi-compartmental model allows PK-PD modeling to account for hysteresis without the PK parameters. This approach may demonstrate an advantage over the full parametric approach when model misspecification is observed in the PK model [15]. Moreover, this semi-compartmental model could be employed in studies where the measured concentration data are difficult to apply to the compartmental PK model, including the PD analysis of inhalational anesthetics using the end-tidal inhalational anesthetics concentration data (PK data) and PD data obtained by electroencephalogram based sedation depth measurements [16, 17].
 XML Download
XML Download