

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Eicosanoids are lipid mediator groups produced by cyclooxygenase (COX), lipooxygenase (LOX), and cytochrome P450 (CYP450) from arachidonic acid. These fatty acids play an important role in all cells, tissues and organs of the body [1]. Eicosanoids can lead to both pro- and anti-inflammatory responses, and the findings of many studies conducted on these response pathways have already been translated into clinical use [2]. Of the three pathways, that involving epoxyeicosatrienoic acid (EET), the metabolite of CYP450, is less clinically known than other pathways [3].
EET blocks inflammation by several mechanisms. First, it inhibits the adhesion of activated monocytes to endothelial cells and reduces tumor necrosis factor alpha (TNF-α), which is an inflammatory cytokine secreted from the by monocytes [4]. Through blockage of NFκB nuclear translocation, EET also downregulates several enzymes, including inducible nitric oxide synthase (iNOS), lipoxygenase-5 (LOX-5), and cyclo-oxygenase-2 (COX-2), which are upregulated during inflammation by blocking NFκB nuclear translocation [5]. EETs are degraded by soluble epoxide hydrolase (sEH) and converted to dihydroxyeicosatrienoic acids (DHET), which is thought to be proinflammatory. Through administration of soluble epoxide hydrolase inhibitor (sEHI), it is possible to prevent the conversion of EET and to antagonize this mechanism of inflammation [6].
The K/BxN T cell receptor transgenic (TCR TG) mouse model, a genetically modified spontaneous arthritis model, was produced by crossbreeding KRN-TCR TG mice with non-obese diabetic mice, resulting in a form of arthritis similar to human arthritis, which was named K/BxN spontaneous arthritis. Histologic examination revealed leukocyte infiltration in joints, synovial cell proliferation, synovitis, and destruction of bone and cartilage were observed, and the mouse was evaluated as a model of acute and chronic arthritis. This K/BxN spontaneous arthritic mouse is complicated to produce, and it is difficult to obtain many individuals for analysis. Therefore, K/BxN serum transfer arthritic mice, in which arthritis is generated through administration of serum from a K/BxN spontaneous arthritic mouse, have been widely used in arthritis experiments [7]. In this K/BxN serum transfer arthritis model, inflammation lasts for several weeks and then disappears. In this model, tactile allodynia and hypersensitivity tend to develop together with the onset of inflammation, then persist for an extended period after resolution of the inflammation [8].
Previously, arthritis had been regarded as an invasive pain associated with the destruction of articular cartilage, and only drugs to treat cartilage destruction had been used. Currently, arthritis is considered to have a neuropathic pain component owing to ischemia, which itself is due to compression and hypoxia; it is also known to be associated with an abnormally excitatory pain pathway in the peripheral and central nervous system [9]. Therefore, anticonvulsant drugs may be used as therapeutic agents. Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit the function of prostaglandin (PG) synthase in the function of COX and reduce the synthesis of prostaglandin E2 (PGE2). Therefore, these drugs inhibit pain and inflammation and are still used, despite various side effects, in acute phase arthritis [10]. Many people are treated with osteoarthritis, and in South Korea, it is estimated that about 10% of the adult population suffers from osteoarthritis [11]. Despite many side effects, drugs such as NSAIDs continue to be used in treatment of this condition. Therefore, we aimed in the present study to investigate the analgesic effect of administering sEHI to stabilize and preserve EET in a mouse model of arthritic pain, and to compare sEHI with existing drugs that have proven efficacy such as diclofenac (DFC) (early arthritis) and gabapentin (late arthritis). We also aimed to observe the presence of synergism between sEHI and DFC using the conditioned place preference (CPP) paradigm, because the withdrawal threshold was not sufficient to observe the analgesic effect.
MATERIALS AND METHODS
Animals
All experiments were performed according to a protocol approved by California State University’s Institutional Animal Care and Use Committee. Male C57BL/6 mice were used in these experiments. The mice were stored in wood chip bedding and plastic cages in a temperature-controlled room and maintained at 12-hour contrast cycle that allowed free access to food and water.
Induction of K/BxN serum transfer arthritis
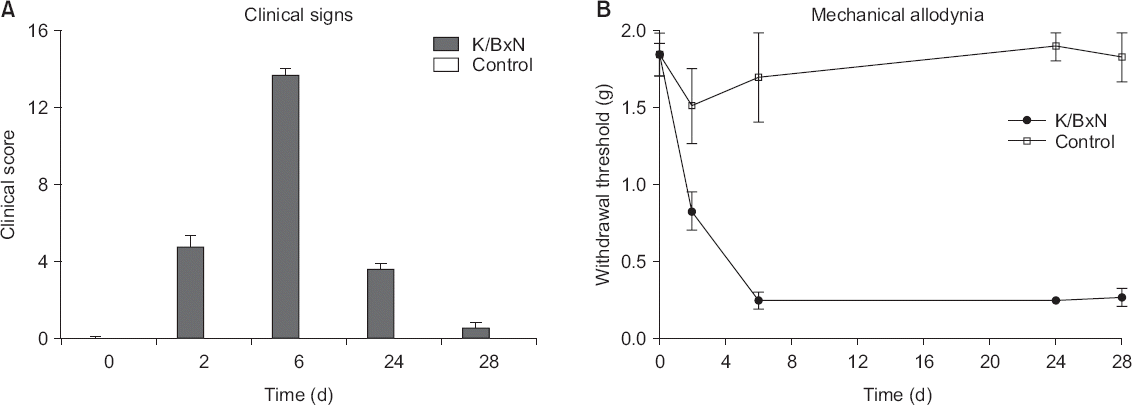
Male C57BL/6 mice (25–30 g) were injected intraperitoneally with 100 µl of K/BxN serum on day 0 and day 2, respectively [7]. Digital signatures such as digital redness and swelling were most prominent on day 6, and the comparison with the control group is shown in Fig. 1. The development of arthritis was assessed by semi-quantitative visual scoring using a scale of 0–4 per paw. Ankle, sole, knuckle, and finger were given 1 point each, with 4 points per each paw and 16 points as the maximum score per mouse [12].
Assessment of mechanical allodynia
All behavioral tests were performed at 9:00 a.m. and 5:00 p.m. at regular intervals. The threshold for mechanical allodynia was measured with von Frey filaments ranging from 2.44 to 4.31. The mice were placed in a plexiglass enclosure on top of the wire mesh surface, and they lived in this environment until 1 hour before testing. Each filament was placed vertically at the center of the hind paw and tested according to the Dixon up-down method [13]. The cutoff value was set at 2 g. The probability of 50% of the withdrawal threshold was calculated and displayed in grams. The mean value of the withdrawal threshold for both posterior paws was measured.
In the early phase of arthritis, sEHI 3 mg/kg, DFC 10 mg/kg, sEHI 3 mg/kg + DFC 10 mg/kg, or saline (vehicle) were administered intraperitoneally, and the analgesic effect was evaluated at 30, 60, 120, and 180 minutes after administration. In the late phase, sEHI 3 mg/kg, sEHI 10 mg/kg, sEHI 30 mg/kg, DFC 10 mg/kg, sEHI 3 mg/kg + DFC 10 mg/kg, sEHI 10 mg/kg + DFC 10 mg/kg, sEHI 30 mg/kg + DFC 10 mg/kg and gabapentin 100 mg/kg, or saline were evaluated. We also examined the synergism between sEHI and DFC using the CPP paradigm.
Conditioned place preference
Conditioned place preference is a standard preclinical behavioral model used to study the beneficial and adverse effects of drugs. The model consists of three compartments with different designs. The center compartment has no special characteristics, and the gates between the compartments can be opened to allow the animal to pass freely. During the training periods, the potentially compensative or aversive material is injected into the animal, and the animal is placed in the specified compartment (not the middle) for a few minutes. The next day, the mice are injected with the vehicle of the drug and placed in the other side compartment. The animal is then placed in the middle compartment in the laboratory; then, both side compartments are opened and a test is performed to record the time the animals spend in each compartment over the test period [14]. We performed this experiment with the hypothesis that reversal of drug-induced allodynia is accompanied by the occurrence of CPP. A modification of this method [15] was used to test the occurrence of CPP for sEHI and DFC in K/BxN serum transfer arthritic mice. Each unit consisted of three adjacent transparent plexiglass compartments.
Each compartment was measured as 90 × 90 × 165 mm, with the center compartment separated by two separate compartments and detachable walls. The middle compartment had no pattern, and the other two sections were a granular vs grid, with one of two textures and either diagonal black-streaked or black square shapes. The total light entering each compartment was the same. The time spent by mice in each room was monitored by recording the collapse of three-stranded light-emitting diode (LED) light paths across the entrance and space of the two patterned compartments. The path of this LED was accumulated by a PC running a data acquisition program based on LabVIEW (National Instruments, USA). The test paradigm was implemented over five days. During the first two days (adaptation day 1 and day 2), the mice were placed in the middle compartment and allowed to move freely between the compartments for 30 minutes. We recorded the time spent in each compartment [16]. In previous experiments [16], mice were placed in the compartment for 30 minutes 10 minutes after the vehicle injection. However, mice were released from the compartment when the maximum effect did not appear after dosing. Twenty minutes after vehicle injection, the mice were placed in one of the two outer compartments for 50 minutes on the morning of the next two ‘conditioning’ days (days 3 and 4). In the afternoon, mice were given medication and after 20 minutes were limited to another external compartment for 50 minutes. On the ‘test’ day (day 5) the mice were placed in the intermediate compartment and were allowed to go in and out of all three compartments freely for 30 minutes; the time spent in each compartment was recorded. To maximize the drug effect, the difference between the time spent in the same compartment on the test day and the average time spent in the outer compartment during the two ‘conditioning’ days was calculated as the outcome. The mice underwent two CPP tests. The test was conducted once in the early stages, and once in the late stages. In each case, mice were randomly administered DFC 10 mg/kg, sEHI 3 mg/kg, or DFC 10 mg/kg + sEHI 3 mg/kg. In the late phase, DFC 10 mg/kg, sEHI 30 mg/kg, or DFC 10 mg/kg + sEHI 30 mg/kg were administered, and the same treatment was not received twice, except for when saline (vehicle) was administered.
RESULTS
K/BxN serum significantly induces arthritis and mechanical hypersensitivity
The K/BxN serum-administered arthritic mouse model showed significant hypersensitivity on day 2 compared to the control group, while significant mechanical allodynia showed the most significant change on day 5, and remained until day 28 (Fig. 1B). The clinical score also began to show on the day 2 and showed the most significant change on the day 6 (Fig. 1A).
Analgesic effect in the K/BxN serum transfer arthritis model induced by sEHI in early phase
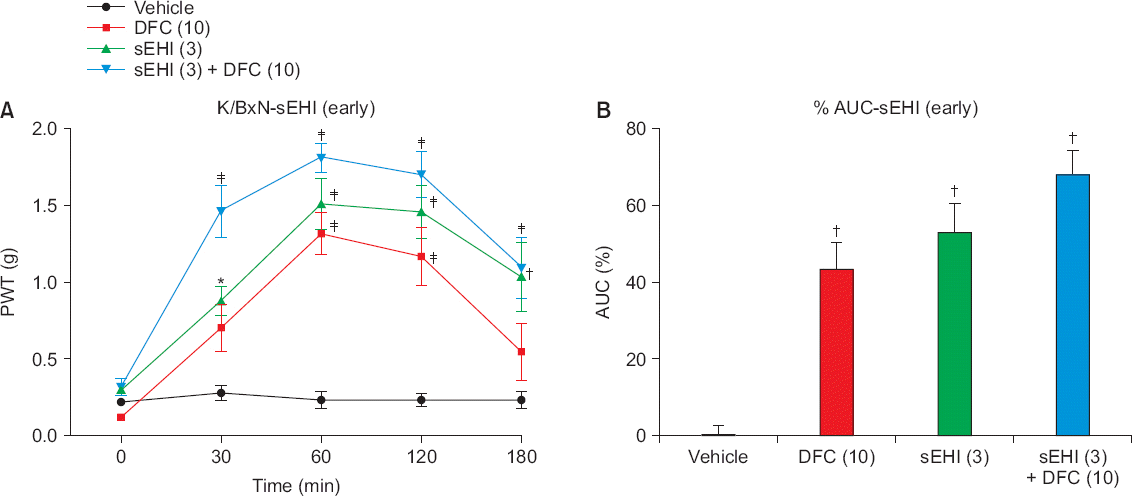
When sEHI was administered intraperitoneally in the early phase of the arthritis model, the paw withdrawal threshold (PWT) increased significantly over 180 minutes (Fig. 2A) and peaked at 60 minutes. This was similar to the findings in the DFC group; DFC was found to be universally effective for early inflammatory reactions. The vehicle-treated control group (n = 6) did not show a significant change in PWT during the same 180-minute test period. In contrast, in the DFC group (n = 6), there was a significant difference only at 60 minutes and 90 minutes (P < 0.001), while in the sEHI (n = 6) and sEHI and DFC groups (n = 6), the difference was significant at 30, 60, 120, and 180 minutes. In particular, the group that received both sEHI and DFC showed the most significant PWT change (P < 0.0001) at all time points. Area under the curve (AUC) analysis shows that DFC, sEHI, and DFC + sEHI increased in order (Fig. 2B). Thus, sEHI showed a greater analgesic effect over the 180-minute test period compared to the control and DFC groups.
Fig. 2
(A) When soluble epoxide hydrolase inhibitor (sEHI) is administered in the early phase of the arthritis model, the paw withdrawal threshold (PWT) increases significantly over 180 minutes and peaks at 60 minutes. This result is similar to the diclofenac (DFC) group. The group that received both sEHI and DFC shows the most significant PWT change (P < 0.0001) at all time points. (B) The area under the curve (AUC) shows that DFC, sEHI, and DFC + sEHI increase in order (*P < 0.050, †P < 0.001, ‡P < 0.0001).
Analgesic effect in the K/BxN serum transfer arthritis model induced by sEHI in the late phase
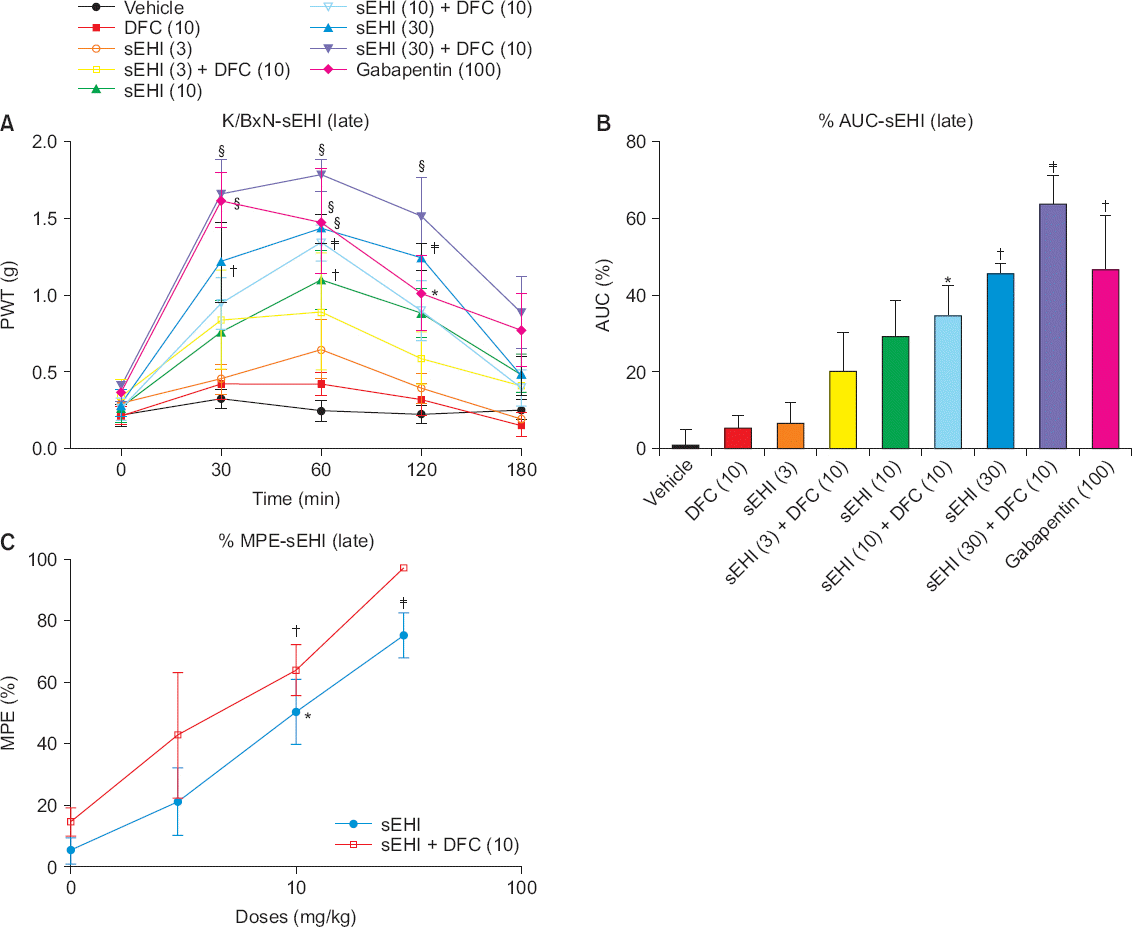
Compared to mice in the early phase, animals in the late phase has been administered more various doses of the drugs. The mice were divided into nine groups; control (n = 4), DFC 10 mg/kg (n = 5), sEHI 3 mg/kg (n = 4), sEHI 3 mg/kg + DFC 10 mg/kg (n = 4), sEHI 10 mg/kg (n = 5), sEHI 10 mg/kg + DFC 10 mg/kg (n = 5), sEHI 30 mg/kg (n = 5), sEHI 30 mg/kg + DFC 10 mg/kg (n = 5), and gabapentin 100 mg/kg (n = 4). PWT was measured at 30, 60, 120, and 180 minutes, as in the early phase, and there was no significant difference among the control, DFC, sEHI 3 mg/kg, and sEHI 3 mg/kg + DFC 10 mg/kg groups (P > 0.050). The sEHI 10 mg/kg group showed a significant difference only at 60 minutes (P < 0.010), but the sEHI 10 mg/kg + DFC 10 mg/kg group showed a more significant difference (P < 0.001) at 60 minutes. The sEHI 30 mg/kg group showed a significant difference at 30, 60, and 120 minutes, with the most significant difference at 60 minutes (P < 0.0001). The sEHI 30 m/kg + DFC 10 mg/kg group showed the most significant difference (P < 0.0001) at 30, 60, and 120 minutes, and higher than in the gabapentin group, even though gabapentin is known to be effective for late arthritis (30, 60 minutes; P < 0.001, 120 minutes; P < 0.050) (Fig. 3A). In the AUC, sEHI 30 mg/kg + DFC 10 mg/kg group had the greatest value (Fig. 3B). In the maximal possible effect (MPE), the sEHI + DFC combination group was more effective than the sEHI alone group (Fig. 3C). Therefore, the combination of sEHI and DFC is effective for treating late arthritis, and sEHI alone is also effective in the treatment of late arthritis.
Fig. 3
(A) In the late phase, no significant difference is shown among the control, diclofenac (DFC), soluble epoxide hydrolase inhibitor (sEHI) 3 mg/kg, and sEHI 3 mg/kg + DFC 10 mg/kg groups (P > 0.050). A significant difference is present in the sEHI 30 mg/kg group at 30, 60, and 120 minutes, with the most significant difference at 60 minutes (P < 0.0001). (B) In the area under the curve (AUC), the sEHI 30 mg/kg + DFC 10 mg/kg group shows the greatest value. (C) In the maximal possible effect (MPE), the sEHI + DFC combination group is more effective than the sEHI alone group over the entire dose (*P < 0.050, †P < 0.010, ‡P < 0.001, ¦P < 0.0001).
Conditioned place preference in the K/BxN serum transfer arthritis model induced by sEHI in both phases
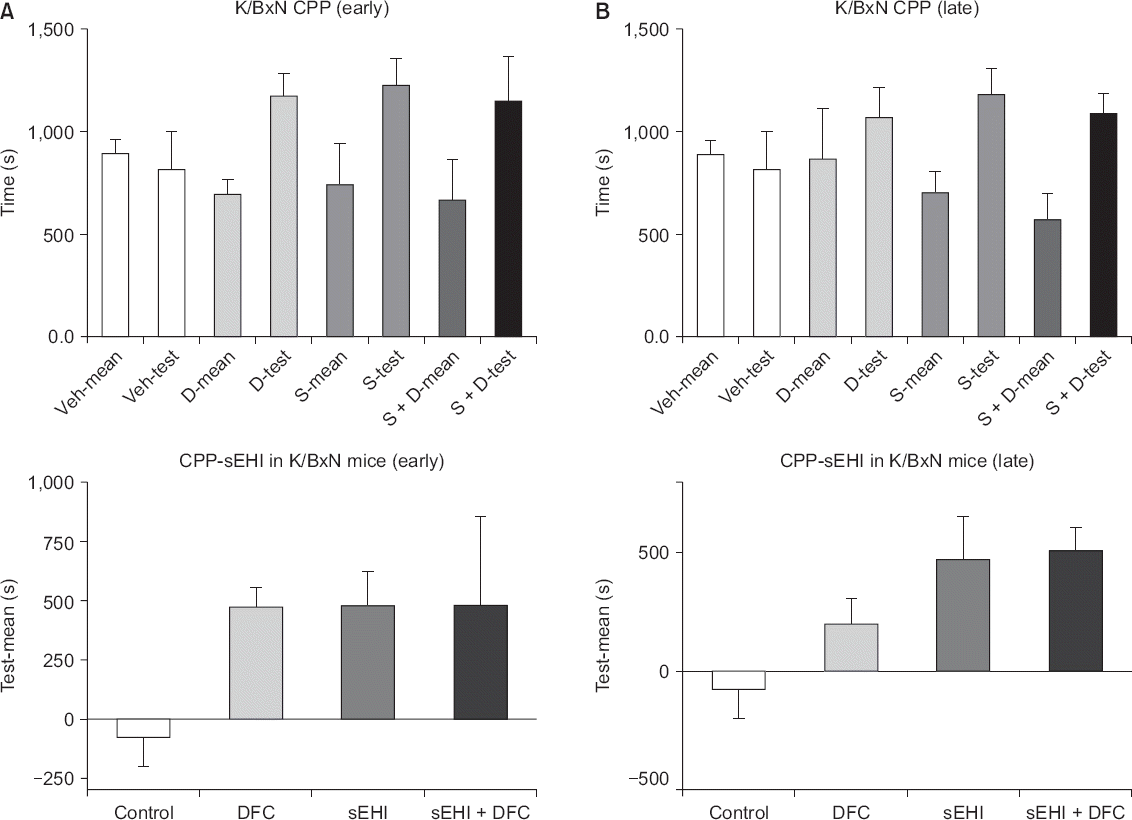
The CPP paradigm test showed that mice favored sEHI and DFC in the early phase and sEHI in the late phase, and showed no synergism was observed between sEHI and DFC in either the early or late phases. In the present study, we tested the place preference of each mouse for each drug and, unlike with the withdrawal threshold assay used previously, we used a single dose. In the early phase, the DFC (n = 4), sEHI (n = 5), and DFC + sEHI (n = 4) groups showed differences in the mean value of the adaptation days and the test days compared to the vehicle (n = 4) group (Fig. 4A). In both the early and late phases, the sEHI and DFC + sEHI groups differed in the mean value of the adaptation day and the mean value of the test day (P < 0.050). The DFC alone group, however, showed no difference compared to the vehicle group in the late phase (Fig. 4B). In both the early and late phases, no synergism was seen in the sEHI + DFC group (Fig. 4).
Fig. 4
(A) In the early phase, the diclofenac (DFC), soluble epoxide hydrolase inhibitor (sEHI), and DFC + sEHI groups show differences in the mean value of the adaptation days and the test days compared to the vehicle group. (B) In the late phase, the sEHI and DFC + sEHI groups differ in the mean value of the adaptation day and the mean value of the test day (P < 0.050). The DFC alone group, however, shows no difference compared to the vehicle group. CPP: conditioned place preference, Veh: vehicle, D: DFC, S: sEHI.
DISCUSSION
This study shows that sEHI is effective in treating early and late arthritis in K/BxN serum transfer arthritic mice. sEHI does not clearly demonstrate a dose-response relationship in early and late arthritis. However, the activity is similar in the early phase to that of DFC, which has been proven to be effective in treating early arthritis. In the late phase, sEHI is more effective than DFC and appears to be more effective than gabapentin, which has been proven effective in chronic arthritis. There is also an additive effect with DFC through CPP, but the existence of synergism is not known.
K/BxN serum transfer arthritis mouse model
The K/BxN serum transfer arthritis mouse model uses antibodies directed to the early, late, and post-inflammatory phases. Unlike the systemic inflammation model, the targeting joint epitope antibody causes only inflammation that is localized to the joints [7]. As is well known, inflammation is associated with allodynia, and even after inflammation is relieved, allodynia can persist through sensitization. As mentioned above, in this experiment, mechanical allodynia started to appear between day 2 and day 5 after the administration of serum, and the clinical score was the highest at day 6, so the early phase progressed to day 9, and after day 28, we considered the inflammation to be completely in remission and we thus proceeded to the late phase experiment on the day 28.
Anti-inflammatory effects of sEHI
sEH is an important enzyme in the metabolism of EET. The EET released from the endothelium has an anti-inflammatory effect, regulates vascular tone, and can play a protective role in the cells. sEHI was first described as a tool for confirming the biological significance of enzymes in the early 1980s [17]. Initially, sEH was found to be present in the liver, kidneys, organs, and blood vessels [18], but it has also been found in neurons as well as the brain [19].
The hypothesis that sEHI can be used to treat arthritis is as follows. First, inhibition of the voltage-activated Na+ channels and enhancement or inhibition of the L-type voltage-activated Ca++ channel have been reported [20]. The relationship between voltage-activated Na channel and pain is well known [21]. The L-type voltage-activated Ca++ channel is not directly involved in peripheral nerves or joints, but it is known to be involved in pain control based on its expression in neuronal cell bodies and dendrites of the dorsal horn [22]. Second, sEHI not only changes the EET metabolite level but also acts on the COX and LOX metabolism pathways. COX is an enzyme well known for mediating inflammatory responses [23]. It is also known to lower the level of inflammation-related substances such as PGE2, leading to anti-inflammatory effects [24]. Third, EET reduces inflammation by inhibiting NFκB-mediated gene transcription and the expression of vascular cell adhesion molecule 1 induced by TNF-α, interleukin-1 alpha, and lipopolysaccharide. In addition, it inhibits PGE2 production and activates transient receptor potential cation channel subfamily V member 1, which has an anti-inflammatory effect, and thus is a basis for treating arthritis [25]. In addition, since EET reduces neuroinflammation and is effective for neuropathic pain, sEHI can also be used in chronic-phase arthritis. This can be explained by the relationship of sEHI to endoplasmic reticulum (ER) stress. In a mouse model with diabetic neuropathy, sEHI has been shown to reduce ER stress and to exert anti-hyperalgesic effects [26].
In general, drugs such as NSAIDs with anti-inflammatory effects have cardiovascular and gastrointestinal complications. However, Norwood et al. [27] have reported that sEHI is associated with no cardiovascular complications such as thrombosis or gastrointestinal complications such as ulcers, and is not toxic when tested at high oral doses in rodents.
sEHI effects for CPP in the K/BxN serum transfer arthritis mouse model
This study demonstrated that sEHI has the ability to treat tactile allodynia in the early and late stages of the K/BxN serum transfer arthritis mouse model and reinforced that CPP has the ability to treat pain. The concept of CPP was originally designed to investigate drug reinforcement, and it is used to test various additive drugs. It is also an alternative for the assessment of pain relief in the absence of stimuli [28]. Clinically, patients with arthritis mainly suffer from spontaneous pain rather than allodynia. Therefore, stimulatory experiments with von Frey are important, but a study such as CPP is helpful to evaluate pain relief in the absence of stimulation. In this experiment, CPP results demonstrated that sEHI had an efficacy in treating arthritis, i.e., inflammatory pain, similar to that of DFC, a medication with proven efficacy for treating early arthritis. In late arthritis, sEHI performed statistically significantly better than DFC for treating arthritis. DFC reversed allodynia in the early phase but not in the late phase, thus initially supporting CPP, but not in the late phase. In terms of site preference, there was statistically significant CPP in the DFC, sEHI, and DFC + sEHI groups compared with the control group in the early phase. In the late phase, only the sEHI and the DFC + sEHI groups showed significant results. As mentioned above, there was no synergism between the two groups. These results demonstrate that sEHI has a positive rewarding effect on arthritis in both the early and late phase by reducing allodynia. Recently, in the CPP paradigm, sEHI has been reported to reverse hyperalgesia in chronic inflammatory pain and improve the withdrawal threshold [29]. In conclusion, in the K/BxN serum transfer arthritis mouse model, sEHI demonstrated similar efficacy to DFC in the early stages of arthritic pain (allodynia), and it was superior to DFC in the late stages, showing gabapentin-like efficacy. The sEHI target thus addresses the hyperalgesia arising from inflammation as well as the post-inflammatory phase that has been said to reflect a neuropathic-like state. Therefore, sEHI offers an alternative to the limited efficacy of arthritis drugs currently in use.
ORCID
JungHyun Park: https://orcid.org/0000-0001-6376-3433
Min-ji Cho: https://orcid.org/0000-0002-2871-6700
 XML Download
XML Download