

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Since neuromuscular blocking agents (NMBAs) were introduced into the surgical field, they have become indispensable for surgery. However, there are many reports of resistance to NMBAs, which are commonly referred to as tachyphylaxis or hyposensitivity [1,2]. Resistance to NMBAs is identified based on increases in the NMBA dosage required to inhibit the muscular twitch response, the time to maximum response, and decreases in the degree of twitch depression or the duration of neuromuscular blockade after a bolus [1].
Approximately 1% of all patients administered general anesthesia exhibit inadequate relaxation, interrupting the procedure [3]. Thus, it is of great importance to understand the mechanisms of NMBA resistance associated with different pathological states to maintain adequate neuromuscular relaxation.
Certain pathological states, such as central nerve injury [4], burns [5], and critical illnesses [6], are associated with resistance to NMBAs. This resistance can be explained by up-regulation of acetylcholine receptors (AChRs) in skeletal muscle [2]. In addition, quantitative and qualitative changes in the physiology of acetylcholine (ACh) and AChR at the neuromuscular junction (NMJ) develop during pathological processes, which lead to changes in the pharmacokinetics and pharmacodynamics of NMBAs [1,2].
PHARMACOKINETIC CHANGES IN NMBA RESISTANCE
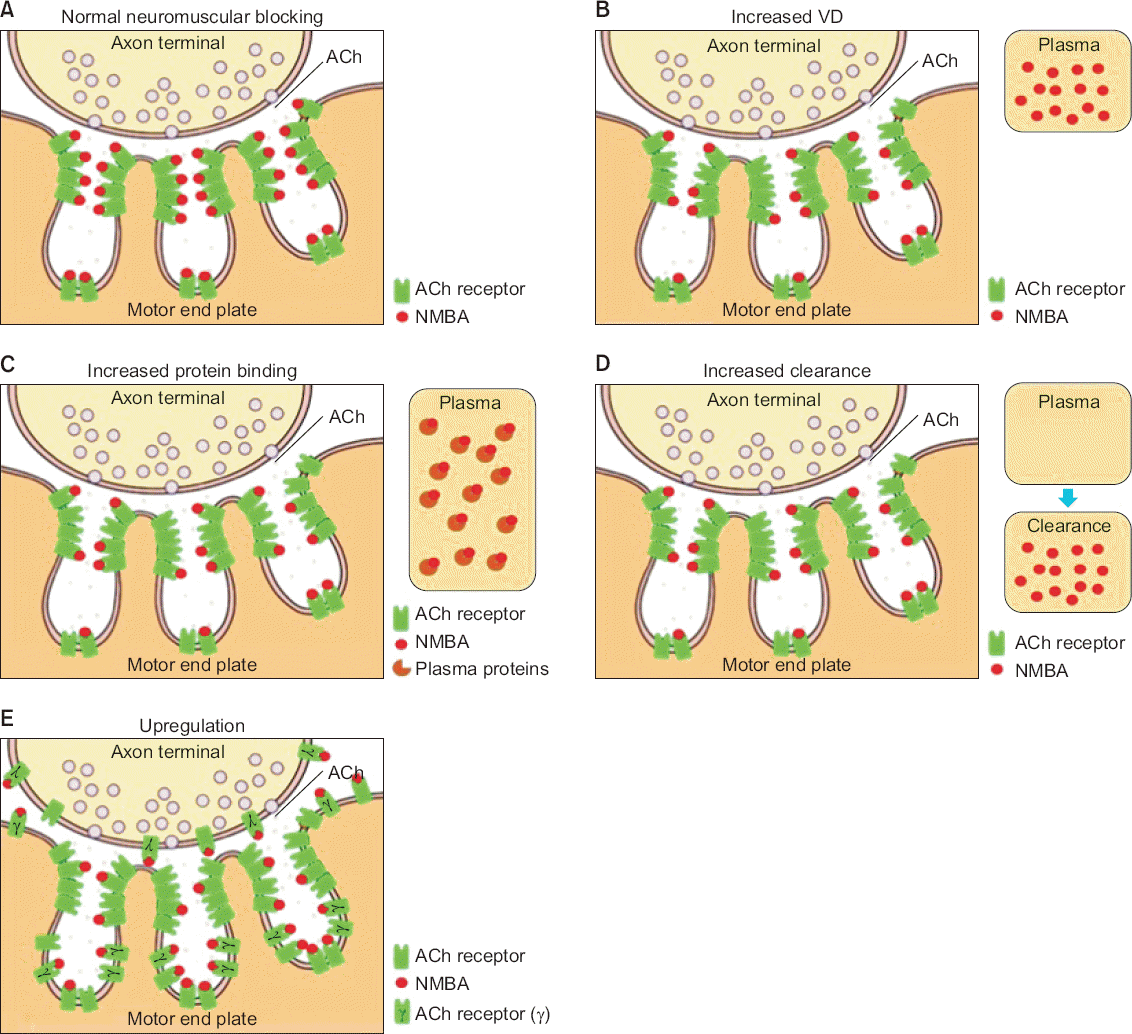
Pharmacokinetic changes in NMBA resistance are associated with changes in the volume of distribution (VD), protein binding, and clearance of NMBAs (Table 1). These changes result in a decrease in the effective NMBA concentration at the receptor site (Fig. 1), and are observed in patients with hepatic dysfunction, thermal injury, oncological diseases, and acid-base disturbances.
Table 1
Pharmacokinetic Changes in NMBA Resistance
Fig. 1
Schematic drawing of the neuromuscular junction (NMJ) and the mechanism of resistance to neuromuscular blocking agents (NMBAs). (A) A normal NMJ and normal interaction between the acetylcholine (ACh) receptor and NMBA. NMBA resistance resulting from pharmacokinetic changes, such as increased volume of distribution (VD), increased protein binding, and increased clearance, is associated with a decrease in NMBA concentration at the receptor site. (B) NMBA resistance associated with increased volume of distribution. (C) NMBA resistance associated with increased protein binding. (D) NMBA resistance associated with increased clearance. Pharmacodynamic changes in NMBA resistance are mostly associated with changes in ACh receptor physiology, such as upregulation, which results in a change in the availability or reactivity of receptors. (E) NMBA resistance associated with upregulation.
Increased VD
An increase in VD may increase resistance to NMBAs, which leads to delayed onset or a shorter duration of action (Fig. 1B). These changes can occur in patients with liver disease, thermal injury, or critical illness. In patients with liver disease, the VD of NMBAs increases [7]; the changes in distribution are multifactorial and depend on the severity of liver dysfunction [1].
Increased protein binding
Diseases, thermal injury, drugs, and acid-base disturbances can increase protein binding of NMBAs and lead to resistance to these agents (Fig. 1C). Usually, acidic drugs bind to albumin and basic drugs bind to α1-acid glycoprotein (AAG) [1]. AAG increases in response to inflammation, surgery, malignancy, myocardial infarction, and thermal injury. An increase in AAG may be responsible for NMBA resistance because the effective NMBA concentration at the receptor site may decrease due to an increase in NMBA protein binding. However, this is only clinically significant when protein binding is >85%. After thermal injury, the plasma concentration of AAG increases and plasma protein binding of NMBAs increases [5,8].
Proteins released from certain tumors are also related to resistance to NMBAs. Patients with adenocarcinoma of the stomach and Wegener’s granulomatosis show a marked increase in AAG and resistance to atracurium because of increased binding to AAG [9,10]. Although, resistance to vecuronium and atracurium has been reported in a patient with multiple myeloma despite a normal AAG concentration [11], paraproteins, immunoglobulin G, and β2-microglobulin increase in these patients, and these proteins bind more NMBA molecules. Thus, availability of NMBA at the receptor site is thought to decrease.
Phenytoin, carbamazepine, and other anticonvulsants may cause resistance to NMBAs [1,2]. Chronic phenytoin therapy decreases recovery time and the recovery index of rocuronium through release of acute-phase reactant proteins such as AAG [12]. However, the mechanisms of phenytoin-induced resistance to NMBAs are complex and include increased hepatic metabolism and clearance through the induction of specific enzymes in the cytochrome P450 system and up-regulation of AChR.
Increased clearance
Increased clearance is also associated with pharmacokinetic changes in NMBA resistance (Fig. 1C). The hyperdynamic state in burn patients, which occurs approximately 48 hours after thermal injury, may increase hepatic blood flow and the glomerular filtration rate, resulting in increased drug clearance [1]. However, pharmacokinetic changes after thermal injury may contribute only partly to NMBA resistance because resistance may continue after recovery from burns [13].
As mentioned above, phenytoin is associated with NMBA resistance through the induction of enzymes in the cytochrome P450 system [12]. Carbamazepine affects NMBA resistance to rocuronium by inducing pharmacokinetic changes, including a two-fold increase in clearance [14].
Resistance to atracurium is related to characteristic metabolism, such as Hofmann elimination and ester hydrolysis. In particular, Hofmann elimination, which accounts for approximately 40% of clearance, is affected by temperature and pH [15]. An increase in body temperature reduces atracurium-induced neuromuscular blockade, and respiratory or metabolic alkalosis also significantly reduce the effects of atracurium and recovery time. However, hyperthermia and alkalosis only appear to contribute minimally to atracurium resistance [1,2].
PHARMACODYNAMIC CHANGES IN NMBA RESISTANCE
Pharmacodynamic changes in NMBA resistance include up-regulation of AChR physiology, enhanced release of ACh at the NMJ, and the inhibition of cholinesterase activity in serum. These changes are seen in patients with denervation injury, thermal injury, immobilization, prolonged use of NMBAs, chronic use of anticonvulsants, and infections (Table 2).
Table 2
Pharmacodynamic Changes in NMBA Resistance
The up-regulation theory refers to a change in the availability or reactivity of receptors (Fig. 1E). In the normal state, AChR consists of five proteins (α, β, ε, and δ in a 2:1:1:1 ratio) only in the junctional area of the NMJ, and the number of extrajunctional AChRs is insignificant [2]. During the absence of neural stimulation or denervation, immature AChRs, with a newly immature glycoprotein γ, instead of ε, develop and proliferate in the junctional and extrajunctional areas of the NMJ [2]. This leads to an increase in the number of remaining unblocked AChRs [16]. In this situation, the effects of a typical dose of an NMBA are weaker, leading to increased sensitivity to agonists and decreased sensitivity to antagonists.
However, not all mechanisms can be explained with these theories, and the pharmacodynamic changes in NMBA resistance are also complex. Other mechanisms have been proposed, such as increased susceptibility of the muscle membrane to depolarization by ACh [17], the effects of NMBAs as partial agonists on immature AChR via altered pharmacological activity [18], and decreased acetylcholinesterase activity after nerve injury [19]. However, the contribution of these components to NMBA resistance is small [2]. The up-regulation and decreased affinity of AChR could be an important component of NMBA resistance.
Denervation injury
NMBA resistance after lower motor neuron injury may be associated with the proliferation of immature AChRs. After such an injury, the number of AChRs increases and resistance to NMBAs occurs only on the injured side [2]. In an animal study in which denervation of the left gastrocnemius was done by creating a 75%–80% lesion of the sciatic nerve, the effective dose of d-tubocurarine increased and the number of AChRs significantly increased in the denervated leg compared to the contralateral leg and uninjured control legs [20]. The effective dose of d-tubocurarine and the number of AChRs were positively correlated.
NMBA resistance develops in patients with an upper motor neuron injury, such as those observed in stroke, cerebral palsy, multiple sclerosis, and hemiparesis secondary to a cerebrovascular accident or cerebral tumor [1,2]. NMBA resistance after a stroke occurs on the paretic side [4] and can start as early as 4–8 hours after the stroke [21]; it is observed most frequently in the distal arm and hand muscles [22]. After an injury, the deprivation of trophic factors or normal input from descending motor pathways lead to central denervation and the transsynaptic degeneration of motor neurons [23]. Furthermore, the number of extrajunctional AChRs increases by collateral reinnervation or axonal nerve sprouting from the surviving lower motor neurons [21].
Thermal injury
NMBA resistance after thermal injury can also explained by denervation-like changes. These changes include fibrillation potentials and positive sharp waves, polyneuropathies and axonal neuropathy, reduced motor nerve conduction, prolonged motor and sensory distal latencies, and reduced amplitude of sensory nerve action potentials [24]. Denervation-like syndrome is associated with increased nicotinic AChR occurrence at the NMJ [25]. In general, 2- to 3-fold higher dosages of NMBAs and 3- to 5-fold higher serum concentrations are required to obtain a general degree of neuromuscular blockage in burn patients [5,26]. A burn with an area greater than 25%–30% of the body surface area that lasts at least 7 days is associated with the development of NMBA resistance [27].
However, the other possible mechanisms of NMBA resistance after thermal injury are complex and associated with other factors, such as immobilization, disuse atrophy, increased protein binding, increase in evoked end plate potentials, altered receptor binding affinity, burn wound-induced contracture, and decreased cholinesterase activity in serum [1,2].
Immobilization and muscle atrophy
NMBA resistance resulting from immobilization and muscle atrophy is also associated with the proliferation of AChR, but it is of a lesser magnitude than denervation syndrome [1,2]. Immobilization does not directly damage the cord or nerves, as muscle fibers are innervated and function normally [19]. The proliferation of extrajunctional AChR, increased ACh sensitivity, terminal nerve sprouting, decreased cholinesterase activity, decreased muscle volume, and decreases in muscle contractile proteins, mitochondria, and sarcoplasmic reticulum have been proposed as etiologies of NMBA resistance based on immobilization and muscle atrophy [2,16]. As has been shown in animal studies, NMBA resistance occurs approximately 4 days after immobilization, whereas an unaffected extremity can show resistance after 1–4 weeks [1,2]. However, the diaphragm is not affected [28].
Prolonged use of NMBAs
The prolonged use of NMBAs may result in resistance through up-regulation of AChR-like immobilization. The chronic use of NMBAs, even in the absence of immobilization or paralysis, causes an up-regulation in the number of receptors and leads to drug tolerance. Chronic administration of d-tubocurarine results in NMBA resistance associated with increased extrajunctional AChR [29]. However, diaphragmatic AChR does not change.
Chronic use of anticonvulsants
Anticonvulsants also cause pharmacodynamic changes in NMBA resistance through antagonism of ACh in pre- and postsynaptic areas [1,2]. The effects of anticonvulsants on NMJ are similar to those of small nonparalytic doses of NMBA. Carbamazepine and phenytoin acutely suppress post-tetanic repetition through their presynaptic inhibitory action on ACh release at the nerve terminal [30]. Therefore, chronic administration of anticonvulsants results in chronic chemical denervation and the subsequent proliferation of AChR.
Infections
Inflammation and infection alters the number of AChRs or the response to NMBAs at the NMJ. The infection-mediated inflammatory response is associated with the release of acute phase-reactant (APR) proteins [8]. NMBAs bind to the APR proteins and then a higher dose is required for neuromuscular block. Toxins from bacteria of the Clostridium genus inhibit the release of ACh at the NMJ; if this state is prolonged, the number of AChRs may increase [31]. Botulinum toxin binds strongly to motor nerve terminals and becomes internalized, which ultimately reduces the release of ACh [31]. Blocking the release of ACh leads to a functionally denervated state in which the muscles became atrophic and extrajunctional AChR dominates.
CONCLUSION
Patients with numerous pathological states are treated with surgery. Some of these patients may be resistant to NMBAs, leading to inadequate neuromuscular blockade, which results in patient movement and interruption of the procedure. Numerous pharmacodynamic and pharmacokinetic changes in VD, protein binding, clearance, and upregulated AChR physiology are associated with the etiology of NMBA resistance. An understanding of the association between the mechanisms of NMBA resistance and the pathological state of the patient would be helpful to maintain adequate neuromuscular relaxation and avoid problems that result from resistance to NMBAs during surgical procedures.
 XML Download
XML Download