

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hearing loss is one of the most common sensorial disorders in children, with incidence rates of one to three per 1,000 newborns [1]. The etiology of hearing loss varies, but at least half of hearing loss cases are due to genetic factors, such as hereditary disorders. About 20% of hearing loss cases in children are associated with inner ear malformations, which may cause profound sensorineural hearing loss [2]. Unsuspected congenital malformations are found in approximately 2.3% of inner ear radiologic studies [3]. Inner ear malformations can result from embryologic arrest, possibly influenced by genetic factors [4].
Lateral semicircular canal (LSCC) malformation is one of the most common radiological anomalies of the bony labyrinth [5]. During the 5th week of embryonic development, superior semicircular canal is the first semicircular canal to develop, followed by posterior semicircular canal. LSCC is the last to develop, and thus may present as an isolated developmental abnormality. The most common form of LSCC anomaly is hypoplasia such as dilatation, as well as shortened or narrowed LSCC [5]. Aplasia, defined as the absence of LSCC, is the most severe form of anomaly and may manifest as part of several broader syndromes, such as CHARGE syndrome [6]. In the literature, Johnson and Lalwani [7] reported that there are diverse types of hearing loss associated with LSCC malformation. The majority of patients with LSCC malformation have sensorineural hearing loss, whereas some have pure or mixed conductive hearing loss. Nevertheless, there have been few reports on vestibular manifestations and performance on audiologic tests in LSCC malformation. Therefore, this study aimed to evaluate audiologic as well as vestibular functions in patients with LSCC malformation.
MATERIALS AND METHODS
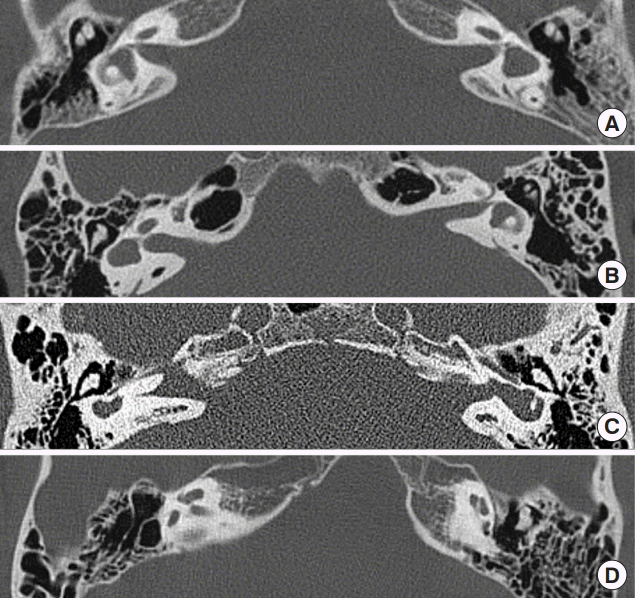
We conducted a retrospective study of patients with LSCC dysplasia at Severance Hospital of Yonsei University Health System, one of the largest tertiary hospitals in Seoul, South Korea. From 2007 to 2017, a total of 15 patients were retrospectively enrolled through the Department of Otolaryngology. Medical histories were reviewed for audiologic symptoms and relevant findings including the etiology of hearing loss, treatment of hearing loss, and accompanying vestibular symptoms. All patients underwent temporal bone computed tomography (CT) scans to evaluate mostly hearing loss, followed by dizziness and chronic otitis media. The enrolled subjects were diagnosed with LSCC dysplasia or aplasia, with or without combined inner ear anomalies, as confirmed by temporal bone CT scans by a radiologist specialized in head and neck section. After that, final diagnosis was made by ENT specialists (Fig. 1). For audiologic assessment, a four-frequency pure-tone average (0.5, 1, 2, and 4 kHz) was obtained from pure-tone audiogram. Vestibular functions were assessed by caloric test and video-assisted head impulse test (vHIT). We calculated canal paresis using caloric test according to Jongkee’s formula. The vHIT of LSCC was analyzed by gain value and the presence of catch-up saccade. This study was approved by Institutional Review Board of Severance Hospital, Yonsei University Health System (IRB No. 4-2015-0659).
RESULTS
Patient characteristics
Fifteen patients (20 affected ears in total) were identified, and their mean age was 34.8 years (Tables 1 and 2). Eight patients were male and seven patients were female. These nine cases were of unilateral LSCC dysplasia (Fig. 1A, B), whereas six patients showed bilateral LSCC dysplasia (Fig. 1C, D). Four out of six patients with bilateral LSCC showed LSCC aplasia, combined with cochlear anomalies. However, one patient with bilateral LSCC dysplasia showed normal cochlear anatomy, although the patient had acquired sensorineural hearing loss on both ears.
Audiologic evaluation
In the unilateral LSCC dysplasia group, the mean average threshold of 0.5, 1, 2, and 4 kHz (PTA4) was 45.5 dB, while three patients had normal hearing levels. In contrast, six patients with bilateral LSCC anomalies had profound hearing loss. Eventually, five patients underwent cochlear implantation surgery (Table 3). Four patients had favorable outcomes after cochlear implantation, as defined by a postoperative Categorized of Auditory Performance (CAP) score of more than 4 points. One patient with comorbid cochlea aplasia had a relatively poor outcome after surgery. No patient complained of dizziness after surgery. One patient showed facial twitching after activation of cochlear implant, which was resolved by decreasing the dynamic range of the offending electrodes.
Vestibular evaluation
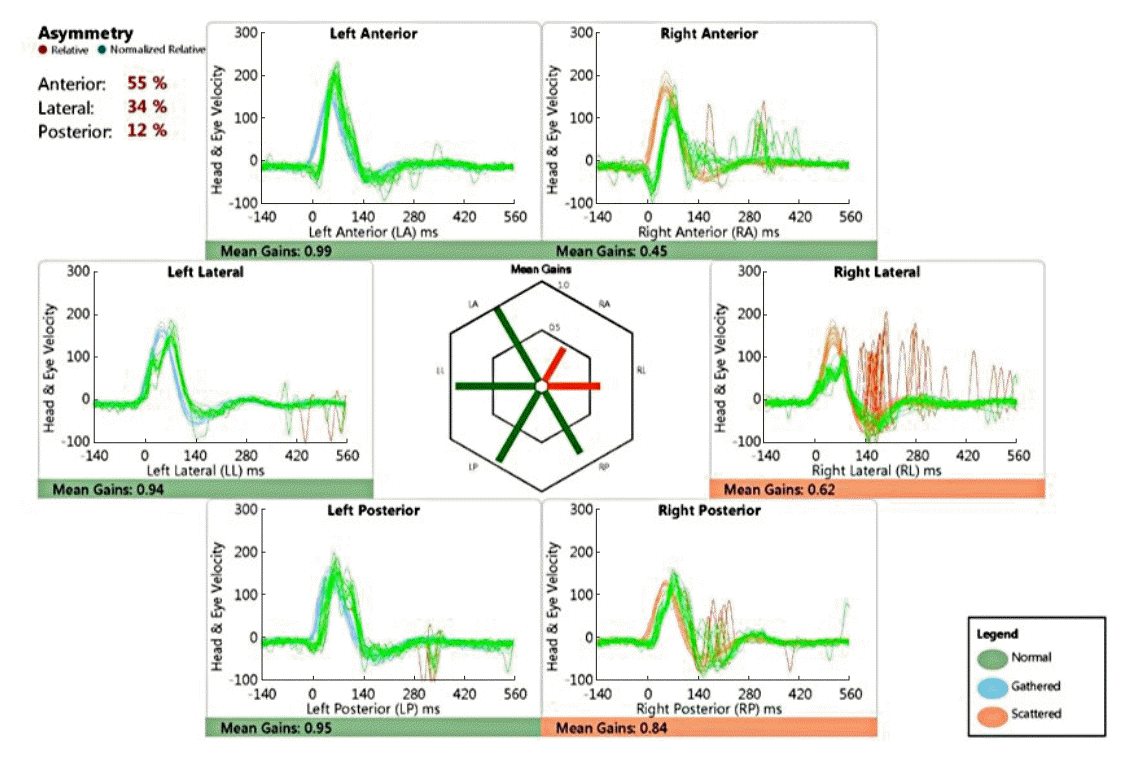
Four out of 15 patients had dizziness symptoms, which were characterized as spinning sensation and/or gait disturbance. Interestingly, dizziness patients all belonged to unilateral LSCC dysplasia groups (Table 4). Two of them described feeling of spinning sensation, whereas the other two patients complained of gait imbalance; their mean age at onset was 37.8 years. All patients underwent caloric stimulation test, and two patients underwent video head impulse test. Only two patients showed normal canal paresis value on caloric testing; two patients with unilateral LSCC dysplasia showed ipsilateral complete canal paresis. Mean canal paresis was 51.8%. In one patient with isolated unilateral LSCC dysplasia (patient 5), horizontal canal gain decreased to 0.62 (17% asymmetry) and anterior canal gain was 0.45 (52.6% asymmetry), whereas posterior canal gain was normal on vHIT (Fig. 2). These results imply that the end organs innervated by superior vestibular nerve are functionally impaired in isolated LSCC dysplasia.
DISCUSSION
In this study, we observed that bilateral LSCC dysplasia or aplasia was usually combined with other inner ear anomalies, resulting in profound bilateral hearing loss and vestibulopathy. Only one patient with bilateral LSCC anomalies showed asymmetric hearing loss. Temporal bone CT of this patient showed more severe combined anomalies on the right side, consistent with the hearing threshold being better on the left side. Therefore, LSCC anomalies combined with other inner ear anomalies resulted in severe hearing loss, in line with previous reports [8,9].
A previous temporal bone anatomy study reported that LSCC anomalies are the most common anomalies of the inner ear [5,10]. LSCC anomalies can be combined with other inner ear anomalies, including those of the cochlea, vestibule, and vestibular aqueduct, depending on which developmental stages of the inner ear are impacted [11]. The semicircular canal develops at embryonic weeks 4 to 7, and early developmental arrest results in deformity of the entire semicircular canal and other structures of the inner ear [12]. Ossification of the labyrinth is completed by the 23rd week, and LSCC is the last of the semicircular canals to complete ossification. Disturbance in ossification may result in LSCC dysplasia, such as lack of a central ossified bony island and a short LSCC complex [13,14].
Our study showed various degrees of hearing loss, with some patients with unilateral LSCC even displaying normal hearing levels. Yamashita et al. [13] reported that there was no correlation with hearing loss in isolated dysplasia of patients with LSCC. Nevertheless, Dallan et al. [15] reported similar results in bilateral LSCC dysplasia. These findings suggest that hearing loss in LSCC dysplasia is likely correlated with anomalous membranous labyrinth development, which we could not find on radiologic examination.
Yu et al. [4] investigated the molecular genetic background of semicircular canal anomalies, and found mutations across a number of genes that resulted in various inner ear defects. During embryonic development, genetic mutations can alter the regulation of transcription factors that play critical roles in the differentiation of specific organs. In addition, Hadrys et al. [16] reported on a knockout mouse model of LSCC malformation in which Nkx5.1, a transcription factor expressed in the presumptive area of the semicircular canal, was deleted. These reports suggest that the pathophysiology of inner ear anomalies may be attributable to genetic defects, and thus genetic counseling should be considered, although the genetic causes of LSCC remain elusive.
The current study suggests a lack of correlation between vestibular function and hearing loss. Only four patients complained about dizziness symptoms, and the onset time of dizziness varied. Different onset times of dizziness symptoms may be attributable to the incomplete compensation by central nerve system and insufficient residual balance function of peripheral vestibular system. Patients with LSCC anomalies are considered to have a small balance capacity; therefore, they are more vulnerable to be affected by uncompenation caused by various origins such as upper respiratory infection infection, malnutrition, stressful condition, or vestibular neuritis.
The results of vestibular examination quite varied according to different patients. Despite the presence of dizziness symptoms, the abnormal value of canal paresis was given on the same side in most unilateral LSCC dysplasia patients. These results indicate that membranous labyrinth had dysplasia, along with bony labyrinth dysplasia, in our patients. In addition, caloric tests were mostly affected by convection effect of endolymph. LSCC dysplasia may change the area ratio in comparison to the normal side, causing canal paresis. McGarvie et al. [17] explained the abnomal canal paresis in caloric test may have resulted from theoretically dissipated hydrostatic pressure in dilated membranous labyrinth in Meniere disease. This theoretical dissipation model can explain our abnormal caloric results on the same side of the affected ear. In contrast, normal values of caloric test in patients with unilateral LSCC dysplasia may infer that anomalies of bony labyrinth are not sufficiently correlated with abnormal vestibular function [18]. As for the results in vHIT, one patient showed normal posterior semicircular canal gain, in addition to abnormal gain in both lateral and superior semicircular canal. This result in vHIT shows impaired superior vestibular nerve in isolated LSCC dysplasia. Superior vestibular nerve innervates to utricle, as well as lateral and superior semicircular canals. Abnormal vHIT and LSCC anomaly can be further explained by the loss of type 1 hair cell. Type 1 hair cells are associated with high acceleration stimuli, which are well correlated with vHIT test. Type 1 hair cells are more abundantly found in crista ampullaris, and LSCC dysplasia might lead to loss of type 1 hair cells and subsequently cause less gain of vHIT [19].
In conclusion, our study showed varying clinical characteristics of LSCC anomalies. Bilateral LSCC dysplasia/aplasia is usually combined with other inner ear anomalies, which may result in profound bilateral hearing loss. However, isolated LSCC dysplasia was associated with varying degrees of hearing loss, and vestibular symptoms did not always accompany LSCC anomalies in our patients.
HIGHLIGHTS
▪ Lateral semicircular canal (LSCC) malformation is one of the most common radiologic anomalies of the bony labyrinth.
▪ Usually bilateral LSCC dysplasia and aplasia may result in profound bilateral hearing loss, due to combined other inner ear anomalies.
▪ Isolated LSCC dysplasia is associated with varying degrees of hearing loss and vestibular symptoms..
 XML Download
XML Download