

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Cisplatin is an important and effective antineoplastic agent that inhibits several types of malignant neoplasms. DNA is believed to be the target of the antineoplastic activity because cisplatin binds irreversibly to DNA, forming intra-strand crosslinks between adjacent guanine residues. Cisplatin has severe side effects that limit its clinical use; these include ototoxicity, nephrotoxicity, and neurotoxicity. Though the mechanism mediating the nephrotoxicity and ototoxicity is unknown, it is believed to be different from that involved in the drug's anti-tumor activity. The most commonly reported histopathological manifestation of ototoxicity is outer hair cell (OHC) degeneration. However, the ototoxic effects of cisplatin are not limited to the auditory hair cells, and the stria vascularis and spiral ganglion cells are also affected (1). Cisplatin-induced ototoxicity is related to changes in the antioxidant activity in the hair cells, particularly the OHCs (2). Mitochondrial dysfunction caused by reactive oxygen species has been implicated in the pathogenesis of cisplatin-induced ototoxicity (3). Boulikas and Vougiouka (4) reviewed that cisplatin directly induced apoptosis in auditory sensory cells through mitochondrial apoptotic pathways. The Bcl-2 family, cytochrome c, and caspase 3 are important elements in the caspase-dependent pathway, and apoptosis-inducing factor (AIF), poly (ADP-ribose) polymerase (PARP), and c-Jun N-terminal kinase (JNK) play critical roles in the caspase-independent pathway. AIF, a mitochondrial intermembrane protein, triggers apoptosis when it is released into the cytosol. Various strategies have been developed in the laboratory setting to prevent ototoxicity, though they are not widely used clinically.
Minocycline is a tetracycline derivative used as an antimicrobial agent to treat conditions such as acne. Many studies have confirmed the anti-inflammatory and neuroprotective properties of minocycline (5). Minocycline inhibits caspase-1 and caspase-3, which are involved in the generation of interleukin-1 and the induction of apoptosis, respectively (6). Wang et al. (7) reported that, in addition to protecting neural cells, minocycline protected non-neuronal kidney epithelial cells against various agents inducing apoptosis through the enhancement of Bcl-2 expression. Corbacella et al. (8) investigated the effect of minocycline on gentamicin (GM)-induced ototoxicity, and reported that minocycline inhibited p38 MAP kinase phosphorylation and caspase 3 activation.
As cisplatin is commonly used for anti-tumor chemptherapy, its ototoxic effect is still a problem for those who suffer from malignant tumors. The aim of the present study was to investigate the protective effect of minocycline on cisplatin induced ototoxicity and to identify the possible mechanism of action in vivo and in vitro.
Go to : 
METHODS
In vitro experiments
Cell culture
House Ear Institute-Organ of Corti 1 (HEI-OC1) cells were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS) at 33℃ under 5% CO2 in an incubator. In all experiments, 70-80% confluent cultures were used.
Cell viability assay
The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay was used to measure HEI-OC1 cell viability after each treatment. HEI-OC1 cells were seeded at 105 cells/mL in 96-well plates and were maintained in DMEM with 10% FBS at 33℃ with 5% CO2 in air. They were exposed to various concentrations of minocycline (Sigma-Aldrich Corp., St. Louis, MO, USA) for 24 hours to determine the proper concentration for the protection experiment. We then selected the 10 µM (4.59 µg/mL) of minocycline for further experiments, which was most protective and less toxic concentration for HEI-OC1 cells (Fig. 1A). Cells were divided into four groups: control, minocycline (mino), cisplatin (cis), and minocycline plus cisplatin (mino-cis). Minocycline was administered 24 hours before cisplatin treatment. Cisplatin was diluted repeatedly using the same volume of media, and the cells were incubated in the various concentrations for 24 hours. MTT solution (2 mg/mL in phosphate-buffered saline [PBS]) was added (50 µL/well) after incubation and plates were further incubated for 2 hours at 33℃ in 5% CO2 in air. The MTT solution was then removed and 100 µL dimethylsulfoxide (DMSO; Kanto Chemical Co., Tokyo, Japan) was added. After stirring the plate using a microplate mixer (Amersham, Pharmacia Biotech, Buckinghamshire, UK), the absorbance at a wave-length of 540 nm was measured using a microplate reader (BIO-RAD 550, Bio-Rad, Hercules, CA, USA). The percentage of cell viability was calculated using the following equation: Cell viability (%)=mean optical density in the test well/mean optical density in the control well×100.
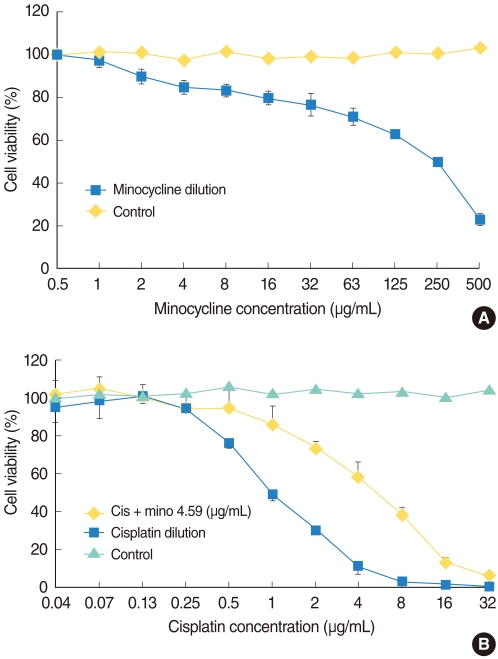 | Fig. 1(A) Cell survival curve of House Ear Institute-Organ of Corti 1 (HEI-OC1) cells cultured with minocycline. Minocyline (mino) is cytotoxic at high, but not at low, concentrations. (B) Cell survival curve of HEI-OC1 cells cultured with cisplatin alone and with cisplatin (cis) after minocycline pretreatment. More cells survived in the 10 µM minocycline pretreatment condition compared with the cisplatin alone condition.
|
Western blot analysis
Following incubation for 24 hours in 10 µM minocycline, 4 µg/mL cisplatin, 8 µg/mL cisplatin, minocycline plus cisplatin (4 µg/mL and 8 µg/mL each) the cells and control plate were analyzed using Western blot analysis. The cells were collected and lysed in radio immunoprecipitation assay (RIPA) buffer (Sigma-Aldrich Co., St. Louis, MO, USA) and the protein concentration was determined using the Bradford assay (Bio-Rad). Protein (100 µg) was loaded onto 12-15% polyacrylamide gels, subjected to electrophoresis, and electrotransferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad). After blocking with 5% nonfat dry milk (in PBS containing 0.1% Tween 20) for 1 hour, the primary antibodies to cleaved caspase-3 (R&D Systems, Minneapolis, MN, USA), Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-c-Jun (Santa Cruz Biotechnology), AIF (Santa Cruz Biotechnology), PARP (Calbiochem, Germany), and GAPDH (Abcam, Cambridge, UK) were applied to the PVDF membranes. The membranes were probed with horseradish peroxidase-conjugated secondary IgG antibody for 1 hour. The proteins were detected by enhanced chemiluminescence (ECL), according to the manufacturer's instructions (Amersham Biosciences). Quantitative analysis of the band density was carried out using Kodak molecular image software (In-vivo FX, New Haven, CT, USA).
Mitochondrial fractionation
AIF is a key protein in the caspase-independent cell death pathway. We measured cytosolic AIF because it is released into the cytosol from mitochondria to trigger cell death. The cells were washed with ice-cold PBS and then resuspended in an isotonic homogenization buffer (20 mM Tris-HCl, pH 7.5, 0.25 M sucrose, 10 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, 1.5 mM MgCl2, 10 µg/mL leupeptin, and 2 µg/mL aprotinin). The cells were incubated in ice for 5 min and microcentrifuged for 10 minutes at 4℃. The supernatant was used as the cytosolic fraction.
In vivo experiments
Animals
We used 15 guinea pigs (200-300 g), free of external and middle ear pathology. All animals had free access to commercial food and water and were maintained in a temperature-controlled environment with a 12/12-hour light/dark cycle. Before the experiment, the animals were anesthetized using an intramuscular injection of a xylazine/ketamine cocktail (ketamine 75 mg/kg, xylazine 5 mg/kg). Deep anesthesia was maintained during the experiment using a half dose of xylazine/ketamine cocktail as required.
The animals were divided into three groups. Group 1 (6 animals) received an intraperitoneal injection (i.p.) of minocycline (45 mg/kg) and received cisplatin (15 mg/kg, i.p.) 12 hours later, group 2 (6 animals) received cisplatin (15 mg/kg, i.p) and normal saline in an equal volume to the minocycline, and group 3 (3 animals) received minocycline (45 mg/kg, i.p) and normal saline in an equal volume to the cisplatin. All chemicals were obtained from Sigma (St. Louis, MO, USA).
Auditory tests
The auditory brainstem response (ABR) was performed using a computer-based signal averaging system (Tucker-Davis Technology, Alachua, FL, USA) to determine the auditory threshold. ABR was performed prior to drug administration and every 24 hours for 5 days after drug treatment. An active electrode lead was positioned subcutaneously at the vertex and referred to the second electrode in the left infra-auricular area. The ground electrode was located on the contralateral ear. ABRs were recorded in an electrically shielded, double-walled sound booth in response to clicks and tone bursts at 8, 16, and 32 kHz. Each response was averaged over a total of 500 times. Intensities were expressed in dB sound pressure level (SPL) peak equivalent, based on calibration. Sounds were presented in 10 dB steps, and ABR responses were recorded for each intensities.ABR threshold was defined as the lowest intensity capable of eliciting a replicable, visually detectable wave V.
Scanning electron microscopy
Immediately after the completion of the follow-up ABRs, animals were decapitated and the cochleae were harvested. Cochleae were immediately immersed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer. Small holes were made in the apex and the round and oval windows. The cochleae were fixed using a perilymphatic perfusion of 2.5% glutaraldehyde in 0.1 M cacodylate buffer and were refrigerated overnight in the same solution. The following day, after rinsing with cacodylate buffer, the cochleae were postfixed with a slow perfusion of 1% osmium tetroxide in 0.1 M cacodylate buffer. The bony capsule of the cochlea was thinned using a dental drill. The remaining bone and lateral wall were dissected away using fine forceps and a 26 gauge needle to expose the organ of Corti. Dissected cochleae were dehydrated in serially increasing concentrations of ethanol from 50-100%, and were critical point-dried. Cochleae were mounted on SEM stubs and were sputter-coated with platinum. Fully processed specimens were examined and photographed using a Hitachi S-3000N scanning electron microscope (Hitachi, Tokyo, Japan).
Statistical analysis
Statistical evaluation of the results was performed using SPSS ver. 10.0 (SPSS Inc., Chicago, IL, USA) with the significance level set at P<0.05. For comparison of the toxicity with serial doses of cisplatin in cell culture experiments, log rank test was used. Mann-Whitney test was used for comparison of hearing before and after cisplatin treatment.
Go to :
RESULTS
In vitro experiments
Cell viability assay
Minocycline was cytotoxic at high, but not at low concentrations (Fig. 1A). Cisplatin was toxic in a dose-dependent manner and pretreatment with minocycline significantly decreased cisplatin-induced toxicity at the concentrations between 0.5 and 16 µg/mL (log rank test, P<0.05). No protective effect of minocycline was observed at higher concentrations of the drug (Fig. 1B).
Western blot analysis
Effect of minocycline on the caspase-dependent pathway: Bcl-2, and caspase 3
To determine whether minocycline inhibited cisplatin-induced caspase activation, cells were exposed to 4 µg/mL and 8 µg/mL cisplatin in the presence or absence of 10 µM minocycline. Cleaved caspase-3 expression increased in HEI-OC1 cells exposed to cisplatin of two different concentrations by 2.22 (4 µg/mL) and 2.32 (8 µg/mL) times as the control. Pretreatment with minocycline attenuated expression of cleaved caspase-3 by half in response to 4 µg/mL, but not 8 µg/mL of cisplatin. The expression of Bcl-2 was elevated by 2.3 (4 µg/mL cisplatin) and 1.98 (8 µg/mL cisplatin) times respectively when minocycline was added to the cisplatin-treated cells (Fig. 2).
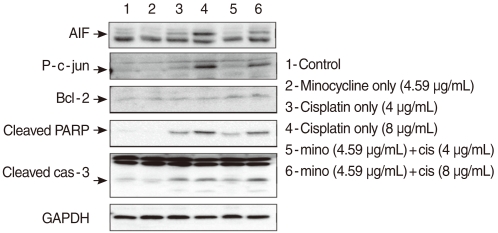 | Fig. 2Cells pretreated with 10 µM minocycline and cultured in 4 µg/mL or 8 µg/mL cisplatin were analyzed using the Western blotting technique with antibodies targeting Bcl-2, p-JUN, cleaved caspase-3, cleaved polymerase (PARP), and AIF. Bcl-2 expression was elevated after the pretreatment with minocycline. Minocycline pretreatment decreased cisplatin-induced cleaved caspase 3 activity at the 4 µg/mL, but not the 8 µg/mL dose. The expression of p-JUN, cleaved PARP, and apoptosis-inducing factor (AIF) increased by cisplatin treatment, and suppressed by minocycline pretreatment.
|
Effect of minocycline on the caspase-independent pathway: JNK, PARP, and AIP
JNK and PARP play important roles in the caspase-independent cell death pathway. To evaluate the role of the pathway in cisplatin-induced apoptosis in HEI-OC1 cells, we measured the expression of p-c-JUN and PARP. The expression of p-c-JUN increased by 3.21 (4 µg/mL) and 15.0 (8 µg/mL) times, in a dose-dependent manner, in the cis group. The expression was markedly attenuated by minocycline pretreatment to the level of 2.26 (4 µg/mL) and 8.04 (8 µg/mL) times as the control. The expression of cleaved PARP also increased by 8.19 (4 µg/mL) and 16.98 (8 µg/mL) times, in a dose-dependent manner, in the cis group. The expression markedly attenuated by minocycline pretreatment to the level of 4.77 (4 µg/mL) and 12.8 (8 µg/mL) times as the control. AIF is a key protein in caspase-independent apoptosis pathway. Cytosolic AIF increased by 2.03 (4 µg/mL) and 1.79 (8 µg/mL) times in a dose-dependent manner in the cis group and decreased to 1.67 (4 µg/mL) and 1.11 (8 µg/mL) times as the control when minocycline was administered (Fig. 2).
In vivo experiments
Auditory brainstem response
No change in hearing was observed in the minocycline-treated animals (group 3). The threshold elevation in group 1 was generally lower than that of group 2. Two animals (33%) in group 1 even did not show any threshold shift. A significant protective effect of minocycline pretreatment was found for the 16 kHz tone (Mann-Whitney test, P<0.05) (Fig. 3).
 | Fig. 3The distribution (A) and mean amount (B) of hearing threshold shift in each group. The shift was greater in group 2 (white circle) than it was in group 1 (black circle). The threshold shift in the 16 Hz tone burst significantly different between the two groups. The threshold shift was less than 10 dB in two animals pretreated with minocycline.
|
SEM evaluation
Morphological evaluation of the SEM sections revealed that the basal and middle turns were more severely damaged than the apical turn. The inner hair cells were relatively well preserved in all turns compared with the OHCs in both groups 1 and 2 (Fig. 4). However, more OHCs survived in group 1 compared with group 2. No morphological change was observed in SEM section of animals treated with minocycline (group 3).
 | Fig. 4Scanning electron micrographs of the basal turn of the organ of Corti. (A) In a hearing-preserved animal pretreated with minocycline followed by cisplatin treatment, the outer hair cells are well preserved with minimal loss. (B) In an animal treated with cisplatin alone, catastrophic destruction occurred in the outer hair cells with fusion of cilia and bulla formation. The inner hair cells are relatively well preserved compared with the outer hair cells in both groups.
|
Go to :
DISCUSSION
It is well known that two mitochondrial apoptotic pathways exist, the caspase-dependent and caspase-independent pathways. Caspase 3, the Bcl family, and cytochrome c (Cyt c) play important roles in the caspase-dependent pathway. PARP-1-mediated cell death is caspase-independent and is considered to be "regulated necrosis" because it lacks many of the morphological features of classic apoptosis. Zong et al. (9) recently reported that in regulated necrosis, the cell actively initiates its death program following PARP-1 hyperactivation. Furthermore, it has been reported that JNK, particularly JNK1, is required for PARP-induced mitochondrial dysfunction inducing AIF translocation and subsequent cell death (10). AIF is an essential downstream element in PARP-1-mediated cell death. It moves into the nucleus and triggers nuclear DNA fragmentation which is an irreversible step in cell death.
Minocycline had been reported to prevent cell death by at least two mechanisms: attenuation of innate and adaptive immunity and blockade of apoptotic cascades (5). Minocycline attenuates immunity through inhibition of microglial activation and proliferation, involving caspase-1, inducible nitric oxide synthetase, and cyclooxygenase 2. Additionally, minocycline inhibits adaptive immunity by reducing the expression of MMPs (11-14). Minocycline inhibits caspase-dependent and -independent cell death induced by different stimuli in vitro, such as glutamate, hydrogen peroxide, and tumor necrosis factor alpha (TNF-α) (15, 16). Up regulation of Bcl-2 produces a protective effect by antagonizing Bax and Bid, a family of death-promoting factors that elicit the release of Cyt c through the outer mitochondrial membrane (7). Alano et al. (17) reported that PARP-1 enzymatic activity can be directly inhibited by tetracycline derivatives and minocycline was the most potent among them. JNK has been reported to have an important role in this apoptotic pathway (10, 18).
To determine the mechanism underlying minocycline protection against cisplatin-induced ototoxicity, we examined cleaved caspase-3, Bcl-2, cleaved PARP, AIF, and JNK, which are important elements in the caspase-dependent and independent pathways.
Cleaved caspase 3 activity was also elevated following exposure to cisplatin. Although addition of minocycline elevated Bcl-2 activity after treatment with both concentrations of cisplatin, it could suppress cleaved caspase 3 activity stimulated only by lower, but not higher concentration of cisplatin. These findings indicate that minocycline does not effectively inhibit caspase-dependent apoptosis stimulated by a high concentration of cisplatin.
HEI-OC1 cells expressed high levels of p-JUN, cleaved PARP, and AIF, which are in the caspase-independent pathway following exposure to 4 µg/mL and 8 µg/mL cisplatin. The increased expression of p-JUN and cleaved PARP decreased by 30-40% with pre-treatment with 10 µM minocycline. The expression of AIF also decreased by 20-40%. These findings suggest that minocycline inhibits PARP activity in the cell nucleus, followed by down regulation of JNK and AIF in the cytoplasm (18) (Fig. 5). Collectively, it can be suggested that cisplatin can induce ototoxicity through two discrete pathways simultaneously, and that these pathways can partially be protected by minocycline.
 | Fig. 5The cisplatin-induced apoptosis pathway in House Ear Institute-Organ of Corti 1 (HEI-OC1) cells and the proposed mechanism mediating the protective effect of minocycline. The black arrow (→) indicates activation, and the blocked arrow head (→|) indicates inhibition. Our data show that minocycline activates Bcl-2 and decreases cleaved caspase-3, poly (ADP-ribose) polymerase (PARP), apoptosis-inducing factor (AIF) activity. These findings and those of previous studies suggest that minocycline activates Bcl-2 and inhibits downstream caspase-3 in the caspase-dependent pathway, and suppresses PARP-1 and downstream AIF in the caspase-independent pathway.
|
The in vivo experiments revealed that the shift in hearing threshold caused by cisplatin administration was partially attenuated by minocycline pretreatment. No change in hearing was observed in two animals of group 1. These results suggest the protective role of minocycline against cisplatin ototoxicity. Though a significant protection was observed only at 16 kHz, this partial effect can be attributed to the small number of animals tested.
Evaluation of the SEM sections revealed that pretreatment with minocycline partially protected the OHCs at the basal and middle turn of the organ of Corti. The morphology of the OHCs was nearly normal in two animals that did not show a shift in the ABR threshold, and the animals with a significant hearing loss showed less severe morphological changes than those treated with cisplatin-alone. The diversity of the protective effect of minocycline may be explained by the difference of drug-host interactions or susceptibility to cisplatin. Moreover, other studies suggest that cisplatin-induced ototoxicity in vivo may be mediated by high-mobility group 1 (HMG1), inducible nitric oxide synthase (iNOS), Na+, K+-ATPase, and Ca2+ ATPase and these various mechanisms of action, not protected by minocycline, may account for the differing responses to minocycline (19, 20).
In conclusion, minocycline may partially protect against cisplatin-induced ototoxicity in vitro and in vivo. Our findings suggest that both caspase-dependent and independent pathways are involved in cisplatin-induced ototoxicity and also in the protective mechanism by minocycline.
Go to :
 XML Download
XML Download