

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
MATERIALS AND METHODS
Genetic screening
Genomic DNA isolation
Polymerase chain reaction amplification and sequence analysis of ENG and ACVRL1
Clinical analysis
Fig. 1.
![]()
RESULTS
Gene variant analyses of Korean patients with suspected HHT
Table 1.
| Gene | No. of families | No. of tested family members | No. of members with genetic variants |
|---|---|---|---|
| ENG | 12 | 35 | 26 |
| ACVRL1 | 12 | 19 | 16 |
| Total | 24 | 54 | 42 |
![]()
Table 2.
| Family no. | Variant location | Variant type | Nucleotide changea) | Protein changea) | Classificationb) | Reference |
|---|---|---|---|---|---|---|
| 1 | Intron 3 | Substitution/splicing defect | c.360+1G>A | p.? | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 2 | 5′-UTR | Substitution/translation defect | c.1-127C>T | New upstream translation start codon | Likely pathogenic (PS3, PM2, PP1, PP4) | [12] |
| 8 | Exon 13 | Deletion/frameshift | c.1687del | p.Glu563Lysfs*10 | Pathogenic (PVS1, PM2, PP4, PP5) | [10] |
| 18 | Exon 13 | Deletion/frameshift | c.1687del | p.Glu563Lysfs*10 | Pathogenic (PVS1, PM2, PP4, PP5) | [10] |
| 20 | Intron 3 | Substitution/splicing defect | c.360+1G>A | p.? | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 21 | Exon 7 | Substitution/missense | c.821C>T | p.Thr274Ile | Uncertain significance (PM2, PP4) | [10] |
| 22 | Exon 1 | Deletion/frameshift | c.63del | p.Thr22Glnfs*21 | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 24 | Exon 3 | Deletion-insertion/nonsense | c.276_277delinsAT | p.Arg93* | Pathogenic (PVS1, PM2, PP4) | This study |
| 26 | Intron 3 | Substitution/splicing defect | c.360+1G>A | p.? | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 31 | Exon 13 | Duplication/frameshift | c.1690_1693dup | p.His565Argfs*3 | Pathogenic (PVS1, PM2, PP4) | This study |
| 33 | Exon 7 | Deletion-insertion/frameshift | c.857_858delinsG | p.Asn286Argfs*73 | Pathogenic (PVS1, PM2, PP4) | This study |
| 34 | Exon 11 | Duplication/frameshift | c.1342dup | p.Leu338Profs*53 | Pathogenic (PVS1, PM2, PP4) | This study |
HHT, hereditary hemorrhagic telangiectasia; p.?, an effect on the protein level is expected, but it is not possible to give a reliable prediction of the consequences; PVS, pathogenic very strong; PM, pathogenic moderate; PP, pathogenic supporting; PS, pathogenic strong; *, termination by nonsense mutation.
a) The reference sequences to describe variants were NC_000009.12 (genomic DNA), NM_000118.3 (coding DNA), and NP_000109.1 (protein).
b) The classification was based on the Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [17].
![]()
Table 3.
| Family No. | Variant location | Variant type | Nucleotide changea) | Protein changea) | Classificationb) | Reference |
|---|---|---|---|---|---|---|
| 5 | Exon 3 | Duplication/frameshift | c.252dup | p.Val85Argfs*84 | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 6 | Exon 3 | Substitution/missense | c.199C>T | p.Arg67Trp | Likely pathogenic (PM2c), PP1, PP3, PP4, PP5) | [10] |
| 10 | Exon 7 | Substitution/missense | c.925G>A | p.Gly309Ser | Likely pathogenic (PM2, PP1, PP3, PP4, PP5) | [10] |
| 12 | Exon 7 | Substitution/missense | c.781G>C | p.Ala261Pro | Uncertain significance (PM2, PP3, PM4) | This study |
| 16 | Exon 10 | Substitution/nonsense | c.1435C>T | p.Arg479* | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 17 | Exon 8 | Deletion/frameshift | c.1118del | p.Lys373Serfs*42 | Pathogenic (PVS1, PM2, PP1, PP4, PP5) | [10] |
| 28 | Exon 3 | Duplication/frameshift | c.121dup | p.Cys41Leufs*128 | Pathogenic (PVS1, PM2, PP4) | This study |
| 30 | Exon 5 | Substitution/missense | c.605T>G | p.Val202Gly | Uncertain significance (PM2, PP3, PP4) | This study |
| 32 | Exon 8 | Substitution/missense | c.1124A>G | p.Tyr375Cys | Uncertain significance (PM2, PP3, PP4) | [10] |
| 35 | Exon 9 | Deletion/frameshift | c.1311_1315del | p.Asp437Glufs*15 | Pathogenic (PVS1, PM2, PP4) | This study |
| 38 | Exon 7 | Substitution/missense | c.1005T>G | p.Asn335Lys | Uncertain significance (PM2, PP3, PP4) | This study |
| 40 | Intron 7 | Substitution/splicing defect | c.1048+5G>A | p.? | Likely pathogenic (PS3, PM2, PP4) | [9,20] |
HHT, hereditary hemorrhagic telangiectasia; *, termination by nonsense mutation; PVS, pathogenic very strong; PM, pathogenic moderate; PP, pathogenic supporting; p.?, an effect on the protein level is expected, but it is not possible to give a reliable prediction of the consequences; PS, pathogenic strong.
a) The reference sequences to describe variants are NC_000012.12 (genomic DNA), NM_000020.3 (coding DNA), and NP_000011.2 (protein).
b) The classification was based on the Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [17].
![]()
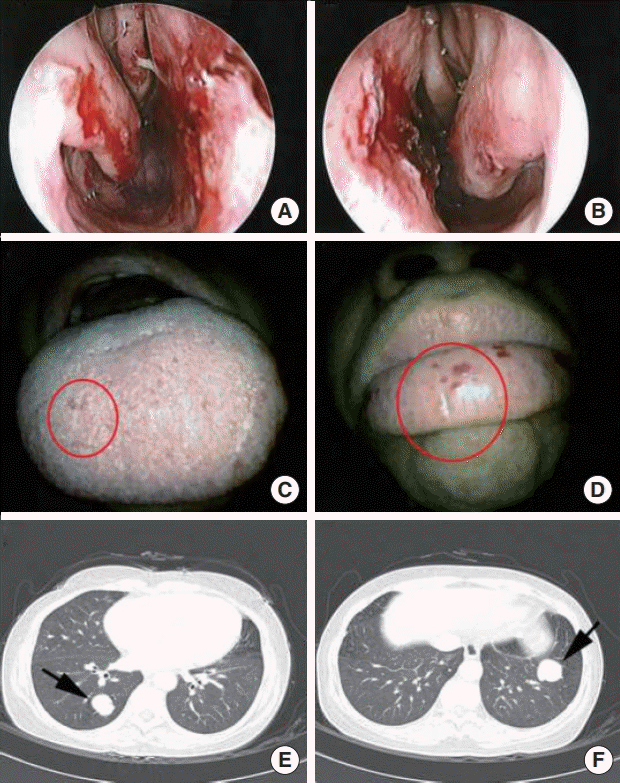
Clinical characteristics of Korean patients with HHT
Table 4.
| Characteristics | Value |
|---|---|
| Sex | |
| Male | 23 (55) |
| Female | 19 (45) |
| Age (yr) | 53.9 |
| Genotype | |
| ENG | 26 (62) |
| ACVRL1 | 16 (38) |
| Clinical presentation | |
| Epistaxis | 38 (90) |
| Telangiectasia | 21 (50) |
| Family history | 37 (88) |
| AVM | 24 (57) |
| Pulmonary | 12 (29) |
| Cerebral | 6 (14) |
| Hepatic | 4 (10) |
| Gastrointestinal | 2 (5) |
| Curaçao criteriaa) | |
| Definite | 24 (57) |
| Probable | 17 (41) |
| Unlikely | 1 (2) |
![]()
Table 5.
| Clinical characteristics | ENG (n=26) | ACVRL1 (n=16) | P-value |
|---|---|---|---|
| Mean age (yr) | 55.3 | 53.4 | - |
| Curaçao criteriaa) | - | ||
| Definite | 17 (65) | 7 (44) | |
| Probable | 9 (35) | 8 (50) | |
| Unlikely | 0 | 1 (6) | |
| Clinical presentation | |||
| Epistaxis | 24 (92) | 14 (88) | 0.628 |
| Telangiectasia | 12 (46) | 9 (56) | 0.751 |
| Family history | 25 (96) | 12 (75) | 0.061 |
| AVM | 18 (42) | 9 (56) | 0.511 |
| Pulmonary | 12 (46) | 0 | <0.001 |
| Cerebral | 3 (12) | 4 (25) | 0.397 |
| Hepatic | 1 (4) | 3 (19) | 0.146 |
| Gastrointestinal | 2 (8) | 2 (13) | 0.628 |
![]()
Table 6.
| Curaçao criteriaa) | No. of members (%) |
|---|---|
| Definite | 6 (20) |
| Probable | 0 |
| Unlikely | 24 (80) |
![]()
 XML Download
XML Download