

 Citation
Citation Print
Print


INTRODUCTION
The correlation of asthma, nasal polyps (NPs), and aspirin intolerance (AI) was first described by Widal et al. [1] in 1922. This full clinical characteristic of AI was identified in the studies of Samter and Beers [2] subsequently, and has been recognized generally as the Samter’s triad (ST). There are many other terms to describe this disease: aspirin-induced asthma, aspirin-sensitive asthma, aspirin hypersensitivity, and aspirin-exacerbated respiratory disease (AERD) including chronic rhinosinusitis (CRS) with as a fourth hallmark of this disease [3].
Although the exact mechanism of ST remains unclear, evidence was found that the pathogenesis of ST is nonallergic hypersensitivity reaction [4-6]. It is associated with the abnormal metabolism of arachidonic acid, which suggest both the lipoxygenase (LO) pathway and the cyclooxygenase (COX) pathway [5,7]. This abnormality causes an imbalance in the synthesis of eicosanoid, prostaglandins (PG) and leukotrienes (LTs). Anti-inflammatory PG, especially E2, decrease and the synthesis of cysteinyl LT (Cys-LT C4, D4, E4) is increased [6-8]. Many patients with ST first visited the otorhinolaryngology department because nasal obstruction and anosmia are the most common symptoms of ST. These patients usually have intractable sinonasal disease that requires multiple endoscopic sinus surgery (ESS).
The provocative challenges test with aspirin or nonsteroidal anti-inflammatory drugs (NSAIDs) is the only diagnostic methods. Aspirin desensitization is an important therapeutic option in patients with ST, especially in those patients suffering from recurrent NPs, anosmia, and overdependence on systemic corticosteroids [5]. This article reviews the recent investigations on the pathophysiological mechanisms and genetic background, diagnosis, and different therapeutic options of ST to advance our understanding of the mechanism and the therapeutic control of ST.
Go to : 
PREVALENCE
It is a difficult to identify the precise prevalence of ST because of the variation in the timing and severity of clinical manifestation of the ST. Furthermore, many patients with ST may not take aspirin or other NSAIDs and may not know the mild reaction due to ingestion [9].
AI is seen in 0.3%–0.9% of the general public, but the prevalence increases to 3%–20% of asthmatics [10,11] and up to 30%–40% in asthmatics with NP [12-14], depending on the populations studied and the diagnostic methods used to evaluate AI. To date, no racial differences have been reported. Many studies show a female predominance [15,16], whereas children younger than 10 years are affected less often since aspirin is contraindicated owing to the risk of Reyes syndrome [17]. Patients with CRS with NP may have more severe AI or asthma than general public. NPs account for up to 70% of AI and asthmatic patients, while the prevalence of the general public is only 4% [16]. The prevalence of ST in patients undergoing ESS has been reported to be 4.8% [18].
Go to :
NATURAL HISTORY AND CLINICAL CHARACTERISTICS
ST is an acquired disease with symptoms beginning from teenager to 40 years old. In most patients, the classic ST develops between the ages of 29 and 34 years [15,16]. About two-thirds of ST patients had positive skin tests to common aeroallergens [15].
ST proceeds gradually from the upper to lower respiratory tract [5,15,16]. Nasal congestion and rhinorrhea are usually the first symptoms, and patients often present a typical viral respiratory infection before the onset of rhinitis. It persists and becomes difficult to treat, and is associated with anosmia, NPs, and recurrent CRS. Anosmia and sinus opacification is found in most patients with ST [15].
Asthma and AI develop 2 to 5 years after the beginning of rhinitis [16]. Twenty percent of patients with ST have a mild asthma, whereas 30% are moderate asthmatic patients who can control with inhaled corticosteroid therapy. Acute asthma attacks usually occur within 3 hours after ingestion of aspirin or NSAIDs and they are usually accompanied by severe rhinorrhea, conjunctival injection, facial flushing, periorbital edema, and abdominal pain. Hypersensitivity reactions induced by aspirin/NSAIDs can occur at any time during the course of the disease. The inflammation of the upper and lower respiratory tract continues even after stopping the use of aspirin and NSAIDs. This supports the fact that intake of aspirin/NSAIDs exacerbates the already active ongoing inflammatory disease than causes the disease for the first time [9].
Go to :
PATHOGENESIS
ST is a non-immunoglobulin E (non-IgE) hypersensitivity reaction associated with aspirin or COX-1 inhibitors, related to an abnormal metabolism of arachidonic acid [3,6,7]. These reactions are associated with an increase in Cys-LTs and eosinophilic cationic protein, reflecting activation of eosinophils and an increase in prostaglandin D2 (PGD2), which mirrors mast cell activation.
Arachidonic acid may be metabolized through COX and 5-LO pathway (Fig. 1). The COX pathway involves the creation of PG and thromboxanes, whereas the LO pathway serves to create LT and hydroperoxyeicosatetraenoic acid. Metabolites produced via the COX pathway under physiological conditions are prostaglandin E2 (PGE2), prostacyclin (PGI2) and thromboxane, and metabolites under inflammatory conditions also include PGD2 and prostaglandin F2 (PGF2). There are two different isoform of COX. Although COX-1 is a continuously expressed enzyme present in most mammalian and most inflammatory cells, COX-2 is an inducible enzyme expressed only in inflammatory cells and upregulated by proinflammatory mediators such as cytokines and growth factors [19]. Arachidonic acid is metabolized to leukotriene A4 (LTA4) via the 5-LO pathway, hydroxylated to leukotriene B4 (LTB4) and converted to leukotriene C4 (LTC4), leukotriene D4 (LTD4), LTE4, leukotriene E4 (LTE4) in the presence of LTC4 synthase (LTC4S) [20].
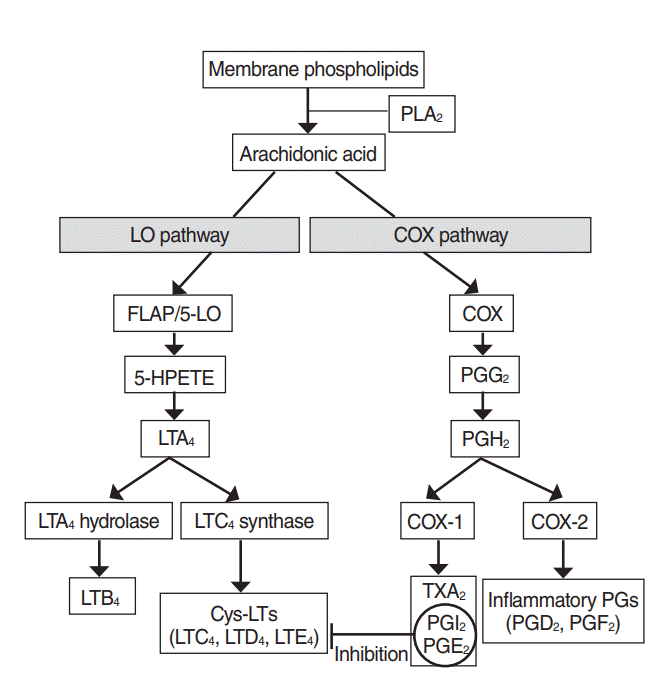 | Fig. 1.Metabolic pathway of arachidonic acid. Via the cyclooxygenase (COX) pathway, prostanoids (prostaglandins [PG], prostacyclins [PGI2], and thromboxanes) are formed, whereas the lipoxygenase (LO) pathway produces leukotrienes (LT). PLA2, phospholipase A2; FLAP, 5-LO activating protein; HPETE, hydroperoxyeicosatetraenoic acid; PGG2, prostaglandin G2; LTA4, leukotriene A4; PGH2, prostaglandin H2; LTC4, leukotriene C4; LTB4, leukotriene B4; Cys-LTs, cysteinyl leukotrienes; LTD4, leukotriene D4; LTE4, leukotriene E4; TXA2, thromboxane A2; PGE2, prostaglandin E2; PGD2, prostaglandin D2; PGF2, prostaglandin F2.
|
COX-1 inhibition is an important event in the development of aspirin-exacerbated respiratory reaction (Fig. 2) [9,21]. By inhibiting the COX pathway, aspirin converts metabolites to the LO pathway, which cause reducing levels of anti-inflammatory PGE2 and increasing the synthesis of Cys-LTs. Moreover, ST patients were known to have an increased LTC4S activity in their bronchial and nasal mucosa [22,23].
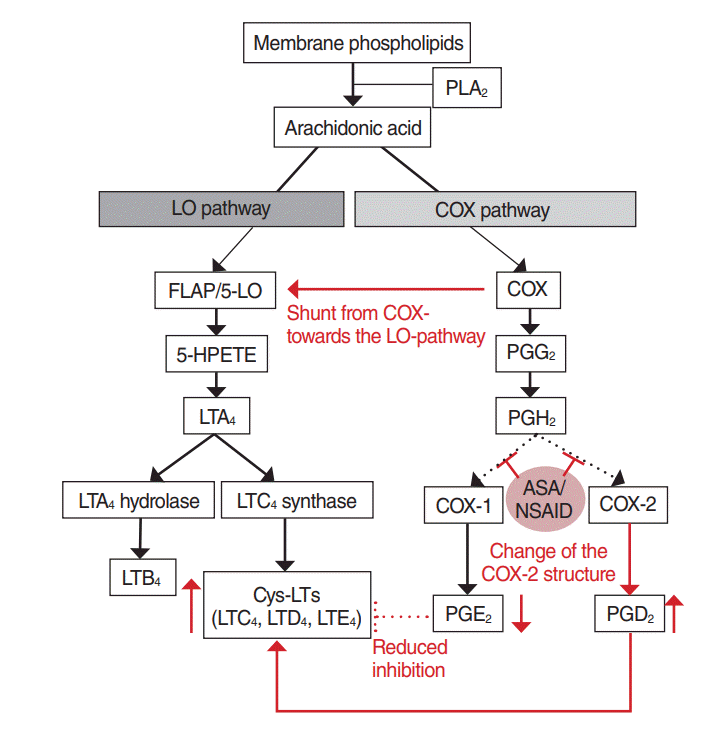 | Fig. 2.Pathogenetic mechanism of Samter’s triad. Aspirin inhibits the cyclooxygenase (COX) pathway and diverts arachidonic acid metabolites to the lipoxygenase (LO) pathway. By inhibition of COX-1, the production of prostaglandin E2 (PGE2) decreases. Low levels of PGE2 lead to increased synthesis of cysteinyl leukotrienes (Cys-LTs). The inhibition of COX-2 might lead to a structural change in the enzyme, which results in the increase of prostaglandin D2 (PGD2). PLA2, phospholipase A2; FLAP, 5-LO activating protein; HPETE, hydroperoxyeicosatetraenoic acid; PGG2, prostaglandin G2; LTA4, leukotriene A4; PGH2, prostaglandin H2; LTC4, leukotriene C4; ASA: acetylsalicylic acid, NSAID, nonsteroidal anti-inflammatory drug; LTB4, leukotriene B4; LTD4, leukotriene D4; LTE4, leukotriene E4.
|
Interleukin (IL)-4 and interferon-γ were increased in the tissues of ST patients. IL-4 is closely related to the upregulation of LTC4S by mast cells [24]. Taken together, these changes in the metabolic pathway of arachidonic acid lead to excessive LT production, which cause eosinophil chemotaxis, increased vascular permeability, mucous gland secretion, and bronchoconstriction. Elevation of Cys-LT levels in the sputum, urine, peripheral blood, and exhaled breath after aspirin administration was observed in previous studies [25]. ST patients had higher levels of nitric oxide and Cys-LTs levels in the urine, blood, sputum and saliva than patients without ST [26].
In addition to increased synthesis of LTs, ST patients have higher Cys-LTs receptor expression in nasal and bronchial inflammatory cells [27]. Although Cys-LTs exert effects only by binding two known G protein-coupled receptors: Cys-LT type I receptor (Cys-LTR1) and Cys-LT type II receptor (Cys-LTR2) [27,28], Cys-LTR1 is only selectively blocked by Cys-LTR antagonist such as zafirlukast, pranlukast and montelukast [29]. In the immunochemical analysis, ST patients showed the higher expression of Cys-LTR1 in inflammatory leukocytes. Therefore, inflammatory cells in ST patients produce more Cys-LTR1, improving their ability to respond to available LT [30]. The recent emergence of molecular biology has revealed a Cys-LT1 receptor polymorphism associated with increased receptor expression in ST patients [31].
PGD2, a mast cell-derived prostanoid synthesized through the COX-2 pathways, is excessively synthesized and secreted in ST patients [32]. PGD2 is a potent chemoattractant of eosinophils that causes vasodilatation and brochocontraction and acts through the PGD2 receptor of eosinophils [33]. Thymic stromal lymphoprotein is thought to play a role in the constitutive overexpression and rapid release of PGD2 during aspirin-induced reaction in addition to its general role as driving T-helper 2 inflammation [34].
PGE2 plays an important role in the pathogenesis of ST. When COX-1 is inhibited by aspirin/NSAIDs, the synthesis of PGE2 is rapidly decreased. Because the “braking” effect of PGE2 diminishes, the synthesis of LTs greatly increased [21,33].
The end result of ST patients is a marked increase in the number of mast cells and eosinophils in the respiratory mucosa. Eosinophils secrete cytotoxic several molecules such as major basic proteins and eosinophilic cationic, causing respiratory mucosal inflammation and injury [9]. Nasal tissue and bronchial biopsy specimens from ST patients showed increased infiltration of degranulated mast cells and eosinophils [35,36].
Go to :
GENETIC BACKGROUND
Recently, several investigations have suggested the problem of genetic polymorphism. Variation within the genes of the arachidonic acid pathway is responsible for changes in the production and metabolism of inflammatory mediators. The association between HLA-DPB1 locus and ST was first introduced [37]. The single allele of DPB1*0301 was overexpressed in ST patients and was suggested as an independent risk factor for ST development [38]. In addition, an association of single nucleotide polymorphisms in genes encoding key enzymes involved arachidonic acid metabolism has been described. Polymorphism in such genes as LTC4S [39], 5-LO [40], PGE2 receptor subtype 2 [41], PGE2 receptor subtype 3 [42], thromboxane A2 receptor [43], and Cys-LTR2 [44] have been revealed to be associated with ST. Recently, IL-10, tumor growth factor-β1, IL-13, and IL-17A gene polymorphism in patients with ST have been reported [45,46].
Go to :
DIAGNOSIS
Taking the history is of primary importance and sometimes diagnostic. Clinician should suspect the ST when patients visit clinic with those conditions or symptoms below (1) a history of attacks of dyspnea or asthma after ingestion of aspirin and other NSAIDs, (2) chronic and intractable nasal obstruction and watery rhinorrhea although allergy skin tests are negative, (3) NPs, (4) pansinusitis in computed tomography, and (5) severe asthma attacks requiring admission to intensive care unit without apparent cause [5-8]. However, the 16% of patients who were clinically suspected to have ST because of a historical asthma attack after ingesting aspirin/NSAIDs cannot meet criteria for ST due to negative aspirin challenges test [14]. On the other hand, of patients who had NPs, CRS, asthma without exposure to aspirin/NSAIDs, only 43% had positive aspirin challenge test [14]. Therefore, ST could to be overdiagnosed (coincidental history) or under-diagnosed (no prior exposure to aspirin/NSAIDs during the time of the inflammatory respiratory disease).
The diagnosis of ST could be confirmed only through the provocative aspirin challenge test by increasing doses of aspirin [47,48]. Four types of provocation test are used according to the route of aspirin administration: oral, bronchial (inhalational), nasal, and intravenous [48]. Oral challenges are most commonly used and regarded as gold standard methods because the oral route mimics natural exposure and the test does not require special equipment [7,48].
Although most ST patients have positive response after taking 30 to 150 mg of aspirin [5,49], the dose should be taken until the cumulative dose of aspirin reaches 500 mg if the patient is not responding according to the EAACI/GA2LEN guideline [50]. Furthermore, if the patient is strongly suspected of having aspirin hypersensitivity, the target cumulative dose can be increased to 1,000 mg [50].
Although not available in the United States, bronchial challenges with L-lysine aspirin have been used in Korea and Europe [48,51,52]. This provocation is safer and faster to perform than the oral route and the symptoms provoked are restricted only to the bronchopulmonary area in most cases. Both provocation tests show similar specificity, but the oral route has higher sensitivity than bronchopulmonary route [48,52].
The nasal challenge is a fast, safe, and less expensive and may be used in outpatient clinic. Although its use is limited by lower sensitivity than oral or bronchial challenge test [53,54], nasal challenge test by a dilute solution of ketorolac has completed diagnostic clinical trials [55,56]. If nasal symptoms are dominant in patients with CRS and NPs, a nasal challenge may be considered as the first-line diagnostic test for ST. Furthermore, only nasal challenge can be available for asthma patients with severe bronchial obstruction [57].
All challenge tests should be preceded by “placebo challenge” to exclude the variability of bronchial responsiveness [21,49]. Aspirin challenge test is considered as positive response when the forced expiratory volume in one second (FEV1) decreased more than 20%, accompanied by clinical symptoms of rhinorrhea, bronchial obstruction, and conjunctival injection. However, severe extrabronchial symptoms without a 20% reduction in FEV1 from baseline can be interpreted as positive response according to the EAACI/GA2LEN guideline [50].
There are no in vitro diagnostic tests for ST although urinary LTE4 has been measured with these provocation tests and elevation of baseline LTE4 synthesis is related with severity of respiratory reactions during oral aspirin challenges [58]. The release of Cys-LTs by peripheral blood leukocytes proved to be nonspecific. However, a recent study reported that fractional exhaled nitric oxide (FeNO) was significantly reduced after 1 hour in 19% of AERD patients after low-dose aspirin (40 mg) challenge [59]. The sensitivity and specificity of detecting FeNO decrease after low-dose aspirin administration were 90% and 100% respectively [59]. Moreover, new noninvasive method such as the assessment of eicosanoid levels in exhaled air condensate or induced sputum and the detection of potential genetic biomarkers could be developed in the near future as interesting diagnostic tools [60]. However, there is no biomarker yet with sufficient sensitivity and specificity to diagnose AERD [61].
Go to :
PREVENTION
ST patients should be educated to avoid aspirin and other cross-reacting NSAIDs that inhibit COX-1 [5,21]. In order to avoid COX-1 inhibitors completely, clinicians should know about cross-reacting NSAIDs drugs that inhibit COX-1 (Table 1). In particular, ST patients must be careful with cough and cold medicines as they often include aspirin or COX-1 inhibitors. Acetaminophen is known as a poor inhibitor of COX-1 and COX-2, but it inhibits a third distinct COX isozyme (COX-3) preferentially [62]. Although acetaminophen inhibits COX-1 only at very high concentrations, it might have cross-reactivity with aspirin when standard therapeutic doses of the drug (650 mg or less) are administrated. However, acetaminophen may induce bronchospasm at dose over than 1,000 mg in 34% of ST patients [63]. Nimesulide and meloxicam, relative inhibitors of COX-2, could induce bronchospasm at higher doses [5]. The highly selective COX-2 inhibitors were known to be well tolerated and could be recommended as safety drug for patients with ST [64-69].
Table 1.
Nonsteroidal NSAIDs that cross-react with aspirin
| Inhibitor pathway | NSAID |
|---|---|
| Predominant COX-1 and COX-2 inhibitors (never take) | Piroxicam, Indomethacin, Sulindac, Tolmetin, Diclofenac, Naproxen, Naproxen sodium, Ibuprofen, Fenoprofen, Ketoprofen, Flubiprofen, Mefenamic acid, Meclofenamate, Ketorolac, Etodolac, Diflunisal, Oxyphenbutazone, Phenylbutazone, Nabumetone |
| Poor COX-1 and COX-2 inhibitors (take only if your doctor agrees) | Acetaminophen (paracetamol), Salsalate |
| Relative inhibitors of COX-2 (take only if your doctor agrees) | Nimesulide, Meloxicam |
| Selective COX-2 inhibitors (okay to take) | Celecoxiba), Rofecoxibb), Valdecoxibb), Etoricoxibc), Parecoxibc), Lumiracoxibc) |
![]()
Go to :
ASPIRIN DESENSITIZATION
Aspirin desensitization is an effective, but not yet commonly used treatment. Aspirin desensitization should be considered as a therapeutic option in patients having intractable respiratory symptoms despite optimal treatment, taking high or chronic oral corticosteroid, and requiring aspirin for the treatment of other diseases [70].
A lot of studies have shown that aspirin desensitization can significantly improve overall respiratory symptoms and quality of life, reduce NP formation and sinus infections, lessen the need of oral corticosteroid and ESS, and improve nasal and asthma symptom scores in ST patients at 6 and 12 months posttreatment [57,71-73]. The necessity for ESS decreased from one operation per 3 years to one operation per 9 years [74].
Another consideration for aspirin desensitization is the timing related to ESS. It is ideal that patients with refractory CRS with NPs should receive aspirin desensitization after ESS. Aspirin desensitization was not effective in reducing existing NPs, but effective in preventing or retarding regrowth of NPs [75]. Although no standard protocol for aspirin desensitization exists, it is usually performed approximately 1 month after sinus and polyp surgery. Aspirin desensitization after ESS was reported to be a well-tolerated and effective adjunctive treatment for long-term control of CRS with NPs in ST patients [76].
Various protocols about aspirin doses and routes of administration are introduced. Although desensitization can be performed only with aspirin or lysine-aspirin [74], the efficacy of nasal challenge using ketorolac opened the way to alternative drugs for desensitization [21,56]. Starting doses and final doses for aspirin desensitization are controversial among prior studies [75,77]. The average threshold doses inducing positive responses after ingestion of aspirin are 45–60 mg. However, the first dose of aspirin starts at 20 or 30 mg because the starting doses for aspirin desensitization should be lower than the threshold doses and aspirin-induced respiratory responses are dependent on the provoking dose.
In the patients who strongly suspected ST but not yet undergone a confirmative aspirin challenge, it would be better to consider a graded challenge as a first phase of the procedure followed by aspirin desensitization. Patients begin with a 20–40 mg oral aspirin challenge, followed by escalating doses of aspirin, progressing through 40–60 mg, 60–100 mg, 100–160 mg, 160–325 mg, and 325 mg, administrated at 90 minute intervals under intensive monitoring. The risk related with severe bronchial reactions during aspirin desensitization will be increased in patients with a history of any previous emergency department visit for asthma when do not use leukotriene-modifying drug at time of challenge and who have baseline FEV1 of less than 80% of predictive value [75].
Therefore, premedication prior to desensitization is important to avoid severe and potentially life-threatening reactions during aspirin desensitization (Table 2). We measure vital signs, FEV1, O2 saturation, and perform naso-ocular, oral, and respiratory assessment every 30 minutes after aspirin administration. If there are ocular and nasal symptoms that stabilize within dose period, we should wait 3 hours before the next dose. We should discontinue desensitization if reaction occurs and the patient’s FEV1 remains under 15% of baseline for more than 3 hours from the last dose despite treatment. Adverse reactions and treatment is summarized in Table 3.
Table 2.
Premedications prior to aspirin challenge and desensitization
![]()
Table 3.
Adverse reactions and treatments during aspirin challenge and desensitization
![]()
Although optimal dose of aspirin for maintenance therapy has not been known [55], generally, high dose aspirin (650 mg twice a day) for controlling airway disease in ST patients is recommended for 1 month. If there is marked improvement in airway symptoms, the dosage was reduced to 650 mg in the morning and 325 mg at night, 325 mg twice a day, and finally 325 mg once a day [76,77]. Although a dose as low as 81 mg of aspirin has been reported to be sufficient for patients requiring cardiovascular prophylaxis, it is usually considered suboptimal for blocking the inflammatory response in the respiratory tract of ST [78]. Therefore, the maintenance dose of aspirin should be at least 325 mg once a day and ideally twice a day [49]. However, recent study showed that daily treatment with aspirin 300 mg has beneficial effects on controlling the upper airway inflammation in ST patients [79]. Furthermore, after successful desensitization, aspirin therapy must be continued for an indefinite time to prevent possible resensitization. Because aspirin desensitization is not an immunotherapy, if the aspirin is discontinued for more than 48 hours, the patients may regain the previous sensitivity within a few days [74,80].
Discontinuation of aspirin is an important barrier to the success of maintenance aspirin therapy. The overall aspirin withdrawal rates ranged from 13% to 46%, and 6% to 18% of patients stopped aspirin due to gastrointestinal complications (abdominal pain, dyspepsia, gastritis or intestinal bleeding) [71,81]. Skin rash, planned surgery, epistaxis, and lack of improvement in respiratory symptoms are other reasons for discontinuation of aspirin therapy. A more recent study reported a lower discontinuation rate of 9% with use of protective medications such as proton pump inhibitors and misoprostol to prevent predictable gastrointestinal adverse events and to improved tolerance [82].
Go to :
MECHANISMS OF ASPIRIN DESENSITIZATION
The pathophysiologic mechanisms of desensitization remain unclear. However, aspirin desensitization leads to decreased LT production, down-regulation of Cys-LTR, and decreased tryptase and histamine release acutely within 3 hours of exposition [83]. After prolonged daily aspirin treatment, the number of nasal inflammatory cells expressing Cys-LTR1 was decreased [7,8,84]. A reduction in urinary LTE4 excretion after 2 weeks and LTB4 synthesis in peripheral monocytes after desensitization has been shown in another study [8].
Direct modulation of intracellular biochemical pathways in key inflammatory cells was reported to another possible mechanism. Daily aspirin therapy inhibited IL-4- and IL-13-induced activation of STAT6 and activation of the transcription factor nuclear factor-κB [85,86]. Another studies have shown that long-term aspirin desensitization involves IL-4 inhibition, downregulation of proinflammatory matrix metallopeptidase 9, an increase in the Th1 marker FMS-related tyrosine kinase 3 ligand, and a decrease in the IL-10 and interferon-γ expression of CD4(+) T lymphocytes [87,88].
Go to :
PHARMACITHERAPY AND SURGERY
CRS in ST has often a serious therapeutic problem. The Samter’s phenotype of CRS is known to be the most severe form of CRS showing higher rates of recurrence after ESS. Antihistamines and nasal decongestants have a limited effect, but LT modifiers [89] and topical steroids [90] are effective in some patients with ST.
Considering the overproduction of Cys-LTs in ST, inhibition with LT receptor antagonist (e.g., montelukast and zafirlukast) and 5-LO inhibitors (e.g., zileuton) would be expected to be effective in ST patients [89,91]. Although ST patients showed more benefit from zileuton than from montelukast, it is less frequently prescribed in ST due to liver toxicity and transaminase monitoring requirement during treatment [92,93].
Monoclonal antibodies can have potential efficacy in AERD. Omalizumab lowered the urinary concentration of LTE4 and PGD2 in patients with AERD and decreased asthma exacerbations and hospitalizations [94]. However, since many patients with AERD have no evidence of specific IgE sensitization, it is difficult for clinicians to prescribe to patients in office and obtain approval insurance guidelines. Benralizumab binding to the IL-5 receptor α induces eosinophil destruction through antibody-dependent cell-mediated cytotoxicity [95,96]. Although specific efficacy for AERD was not addressed, benralizumab is effective in patients with eosinophilic asthma [96,97]. Since the expression of eosinophils increases in patients with AERD, the target therapies of IL-5 may be effective, but specific studies are needed to confirm efficacy in this population.
The outcome of ESS in patients with ST has previously been reviewed. Many studies supported the role of surgery to improve sinonasal symptom severity and frequency, radiologic and endoscopic scores, quality of life, and asthma severity [76,98-105]. Most ST patients showed improvement in asthma severity scores, decreased emergency room visits and hospitalization, and reduced need for inhaled and oral steroid use after ESS [105-107]. However, ESS clears gross NP burden but does not address the underlying abnormal metabolism of arachidonic acid. Although subjective success rate of ESS is estimated up to 80%, many patients continuously complain of nasal symptoms such as nasal obstruction and anosmia over time. According to the follow-up duration, recurrence rate of NPs is reported at least 40%. Therefore, it is ideal for patients with ST to receive additional treatment like aspirin desensitization following ESS to prevent recurrence of CRS with NPs.
Go to :
IMPLICATIONS FOR PRACTICE
ST is a pseudoallergic non-IgE-mediated hypersensitivity reaction associated with an abnormal metabolism of arachidonic acid and COX-1 inhibition is the key event. Aspirin desensitization reduces nasal obstruction and polyp regrowth, improves respiratory symptoms, and consequently reduces the need for continuous medication and the need for surgery. More patients with ST can be successfully managed by a greater awareness of ST and the effects of aspirin desensitization.
Future research should focus on identifying biomarkers for early diagnosis based on various diagnostic techniques. A better understanding of the genetic background, molecular, cellular and biochemical basis of ST will help to determine new diagnostic tools and therapeutic interventions.
Go to :
HIGHLIGHTS
▪ Samter’s triad (ST) is nonallergic hypersensitivity reaction.
▪ We review the pathophysiology, diagnosis, and therapeutic options of ST.
▪ Aspirin desensitization reduces nasal symptoms and the need for continuous medication and surgery.
▪ The patients with ST should receive aspirin desensitization after endoscopic sinus surgery.
Go to :
 XML Download
XML Download