

 Citation
Citation Print
Print


INTRODUCTION
Tobacco smoking is an important environmental health problem that is associated with many human diseases [1]. Maternal smoking increases the risk of various complications, ranging from miscarriage to fetal brain development [2]. Among more than 7,000 chemicals in tobacco, nicotine is undoubted one of the most widely studied agents. Nicotine acts through the stimulation of nicotinic acetylcholine receptors (nAChRs) located on neuronal or non-neuronal tissues [3]. In addition, these receptors are represented during development, and prenatal maternal nicotine exposure results in diverse adverse effects in various animal models [2,3].
Zebrafish (Danio rerio) is known to be an effective model for the research of embryotoxicity because of its ex utero embryo development, transparency of embryos, and short seeding period [4,5]. Since nAChRs are confirmed to be represented in zebrafish embryos, and regulate nicotine-induced changes in embryonic morphology, zebrafish are appearing as a useful model for conducting the molecular and biological effects of nicotine exposure [5-7]. In zebrafish, embryonic nicotine exposure results in various development problems including growth retardation [8], altered neural development [5], cardiac toxicity [9], muscle development alteration [10], and behavior abnormality [2,5].
Although data on the association of smoking and hearing loss have been in contrast to human adult cohort studies [11,12], smoking was known to deteriorate the auditory thresholds at high frequencies [13] and cause lower response levels in transient evoked otoacoustic emissions [14]. In particular, prenatal or childhood nicotine exposure affects hearing in humans [14-17]. In addition, nicotine administration in adult guinea pigs caused damage of outer hair cells, especially at the stereocilia, and expansion of the surrounding supporting cells [18]. However, the hair cell damage by prenatal nicotine exposure has not yet been entirely established until recently. In the current study, we assessed the in vivo hair cell toxicity and embryotoxicity of nicotine during zebrafish development.
Go to : 
MATERIALS AND METHODS
Chemicals and zebrafish preparation
The (–)-nicotine (product no. N3876) used in this study was acquired from Sigma-Aldrich (St. Louis, MO, USA). Nicotine solutions were prepared in distilled water and diluted in embryo medium to achieve final concentrations. In addition, this solution was produced daily as needed for all experiments.
Wild-type (AB) and transgenic (Tg; brn3c:EGFP and cmlc2: EGFP) zebrafish were sustained at 28.5℃ under a 14 hours light and 10 hours dark cycle in a zebrafish facility at Korea University Ansan Hospital as previously described [7,19]. Newly hatched brine shrimp (Artemia; San Francisco Bay Brand Inc., Newark, CA, USA) were supplied two times per day for the adult fish. Tg (brn3c:EGFP) and Tg (cmlc2:EGFP) express green fluorescence characteristically in hair cells and myocardial cells, allowing evaluation of heart size, respectively. The wild type and Tg zebrafish larvae were produced by paired matings of age-matched adult fish, and larvae were sustained at a density of approximately 50 embryos per 100-mm petri dish in embryo media as previously described [20].
Our study was permitted by Institutional Animal Care and Use Committee of Korea University Ansan Hospital (approval No. KUIACUC-2015-173). All tests were performed within the guidelines of the Animal Care Ethics Committee of Korea Universityand the National Institutes of Health guidelines.
Assessment of embryotoxicity in zebrafish
Larvae (wild-type AB strain) were seeded in a 6-well plate (5 mL volume for each well) with different concentrations of nicotine (0, 5, 10, 20, and 40 μM) and then incubated at 28.5℃±1℃ for 72 hours (10 larvae per well; 80 larvae per treatment concentration; total n=400). Because the stability of nicotine in embryo media is not determined, the nicotine-containing embryo media was replaced daily. All assessments of embryotoxicity were performed at 72 hours post-fertilization (hpf) using microscope (LUMAR V12, Carl Zeiss, Jena, Germany; 6.4–80× magnification, AxioVision 4.8.2, Carl Zeiss) as previously described [4]. The hatching rate and the mortality rate were calculated from the number of hatched larvae and the number of dead larvae divided by the number of total larvae, respectively. Teratogenicity rate was counted from number of fish showing systemic teratogenicities including bending of the spine and tail, stunted growth, malformed yolk sacs, and edema in the body cavity. In addition, the rate of total abnormal findings (mortality rate+teratogenicity rate) was also demonstrated. For evaluation of heart malformation, Tg (cmlc2:EGFP) zebrafish morphology was checked at 72 hpf. Also, heart rate at 72 hpf was scored real-time by direct microscopic inspection for 60 seconds at each concentration of nicotine (n=30 for each group), while larvae were anesthetized with tricaine (200 μL).
Assessment of hair cell toxicity in zebrafish
The wild-type and Tg (brn3c:EGFP) larvae were revealed to nicotine at 5, 10, 20, and 40 μM for 120 hours. At 120 hpf, the larvae were rinsed with embryo medium three times and anesthetized with a tricaine (3-aminobenzoic acid 0.4 g/ethyl ester; 100 mL; pH 7, adjusted using Tris buffer) for 5 minutes as previously described [19].
In the Tg (brn3c:EGFP) larvae, hair cells in four neuromasts (supraorbital [SO1 and SO2], otic [O1], and occipital [OC1]) were analyzed on one side of each fish under a fluorescence microscope (AxioCam MRc5, Carl Zeiss; 5, 10, 20, 40× magnification, AxioVision 4.8.2). The total number of hair cells within four neuromasts (three times replicated with n=30 for each group) was analyzed. Furthermore, apoptosis of hair cell was identified at 120 hpf in Tg (brn3c:EGFP) zebrafish by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining with an in situ cell detection kit (Roche Molecular Biochemicals, Mannheim, Germany) in accordance with the manufacturer’s protocol. TUNEL-positive cells were analyzed in four neuromasts in 10 fish from each group (total n=50), as previously described [19].
To evaluate mitochondrial damage within the hair cells, the fluorescent dye, 2-(4-[dimethylamino]styryl)-N-ethylpyridinium iodide (DASPEI; Invitrogen, Carlsbad, CA, USA) was applied to wild-type larvae. After anesthetization, larvae were incubated in embryo medium containing 0.005% DASPEI for 15 minutes. The DASPEI-stained area within SO1, SO2, O1, and OC1 was examined under a fluorescence microscope and measured using Image J (ver. 1.48; National Institutes of Health, Bethesda, MA, USA) as we previously described [20]. The average area of DASPEI staining in neuromasts was compared among all experimental groups and the control group (n=6 fish for each group).
Measurement of intracellular reactive oxygen species generation
To examine whether the reactive oxygen species (ROS) production is influenced by nicotine exposure, we evaluated intracellular ROS generation using an oxidation-sensitive fluorescent probe dye; a 2,7-dichlorodihydrofluorescein diacetate (DCHF-DA). Wild-type zebrafish were washed three times with embryo media, and seeded into 24-well plates. And then, DCHF-DA was treated with 10 μM for 5 minutes, and washed three times in embryo media. For DNA staining, 4´,6-diamidino-2-phenylindole (DAPI) was applied for 3 minutes in 0.1 mg/mL. After washing again, groups were subdivided for normal control, positive control (125 μM neomycin for 20 minutes), and two experimental groups treated with 5 μM and 40 μM nicotine (for 20 minutes). After the treatments, larvae were anesthetized and mounted in 2% methyl cellulose for observation under a fluorescence microscope.
Scanning electron microscopy
The 5-dpf wild-type larvae exposed to 5 μM and 40 μM nicotine and normal controls were prefixed by immersion in 2% glutaraldehyde in 0.1 M phosphate buffer and postfixed for 2 hours in 1% osmic acid dissolved in phosphate-buffered saline. Larvae were managed in a graded series of ethanol and t-butyl alcohol, dehydrated in a freeze dryer (ES-2030; Hitachi, Tokyo, Japan), platinum coated using an ion coater (IB-5; Eiko, Tokyo, Japan) and investigated using scanning electron microscopy (SEM; S-4700; 2,500, 3,000, 5,000× magnification, Hitachi) as previously described [20].
Statistical analysis
Statistical comparisons were performed using likelihood ratio test for trend for categorical data including hatching rate, mortality rate, teratogenicity rate, and total abnormal findings. The one-way analysis of variance (ANOVA) was used for multiple comparisons of numerical data and a post-hoc analysis was performed using Tukey’s honestly significant difference test. In the current study, statistical analysis was conducted with IBM SPSS ver. 20.0 (IBM Corp., Armonk, NY, USA). A P<0.05 were considered statistically significant.
Go to :
RESULTS
Embryotoxicity of nicotine in zebrafish (brn3c and cmcl2)
The control group without nicotine exposure appeared normal with an overall mortality and teratogenicity rate <5%, and hatching rate of 96% (Fig. 1). The hatching rate was not significantly different according to nicotine concentration (P=0.216, linear by linear association). At 72 hpf, 81% of embryos hatched in the 5 μM nicotine group compared with 84% in the group treated with 40 μM (Fig. 1). The mortality rate was also not significantly different between the normal control and nicotine-treated groups (P=0.637, linear by linear association) (Fig. 1). The mortality rate was 1%, 15%, 19%, 19%, and 9% in control, nicotine 5, 10, 20, and 40 μM groups, respectively. With increasing nicotine concentration, the teratogenicity rate markedly increased (P<0.001, linear by linear association) (Fig. 1). The teratogenicity rate was 1% in the 5 μM nicotine group and 44% in the 40 μM nicotine group. Furthermore, the rate of total abnormal findings notably increased in a dose-dependent manner (P<0.001, linear by linear association) (Fig. 1). The results of embryotoxicity for different treatment concentrations of nicotine are presented in Fig. 1.
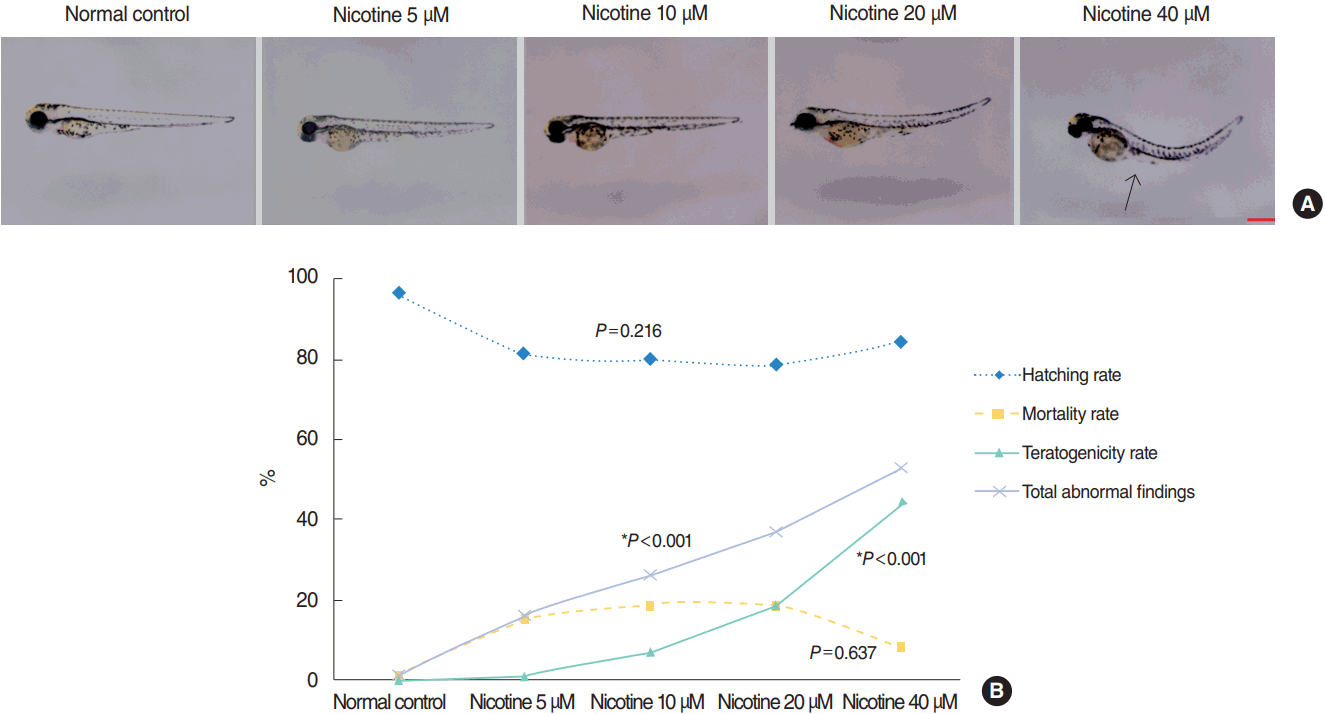 | Fig. 1.Nicotine-induced embryotoxicity at 72 hours post-fertilization (hpf). (A) Hatching/non-hatching embryo findings. The number of embryos showing abnormal morphology tended to be increased in the nicotine-exposed groups (arrow: bending of the trunk). All images were captured at 72 hpf (×32). Scale bar=1,000 μm. (B) Hatching rate, mortality rate, teratogenicity rate, and total abnormal findings (mortality rate+teratogenicity rate) showed statistically significant differences among nicotine concentrations (n=100 per concentration, linear by linear association). Nicotine-exposed groups showed a lower hatching rate and higher mortality and teratogenicity than the control group. The rate of teratogenicity and total abnormal findings increased in a dose-dependent manner (1% in the normal control group, 16% for 5 μM nicotine, 26% for 10 μM nicotine, 37% for 20 μM nicotine, and 53% for 40 μM nicotine; (total n=400). *Statistically significant. All data were evaluated at 72 hpf.
|
No significant heart malformation was observed in nicotine-exposed embryos of Tg (cmcl2) (Fig. 2A). The heart rate significantly decreased as nicotine concentration increased (P<0.001, n=30 for each concentration, ANOVA) (Fig. 2B).
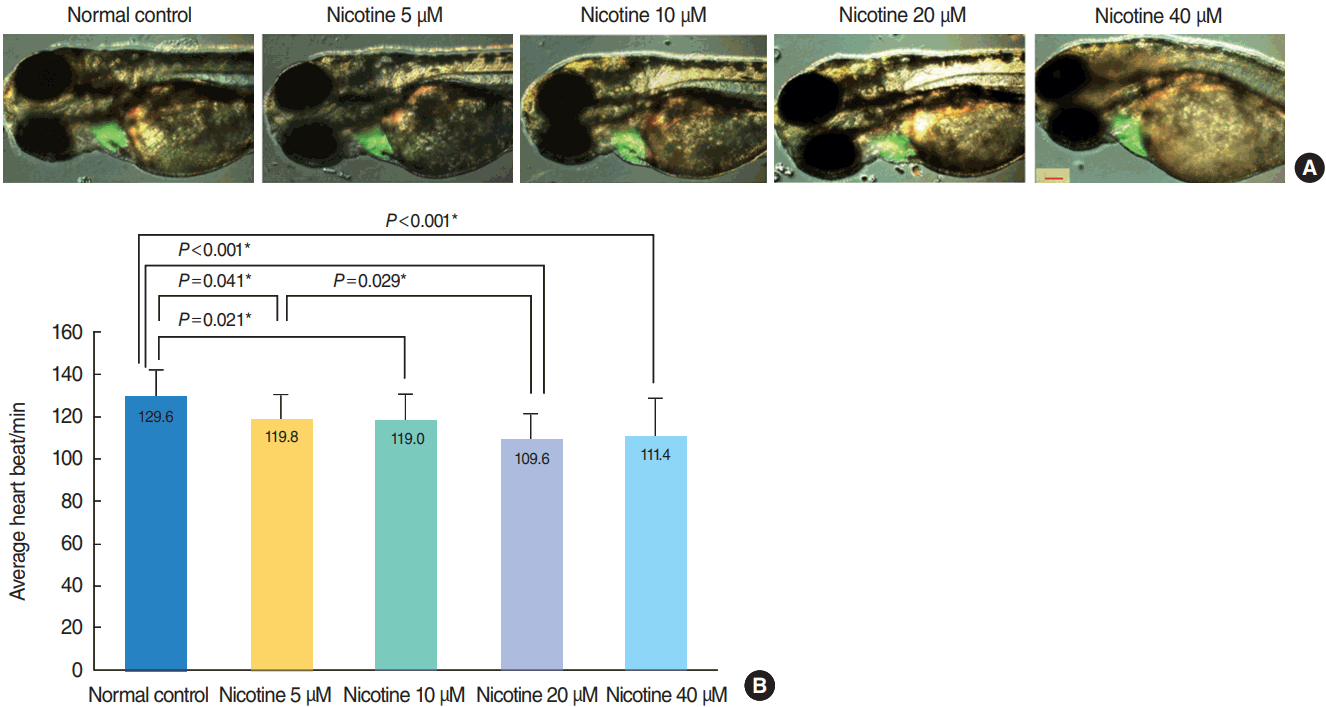 | Fig. 2.Heart morphology evaluation and changes in heart rate induced by nicotine treatment. (A) Merged images from the fluorescent image and optical image are shown. There was no difference in heart size between groups (green area). All images were captured using transgenic (cmlc2:EGFP) zebrafish at 72 hours post-fertilization (hpf, ×16). Scale bar=200 μm. (B) The heart rate of zebrafish embryos was significantly decreased in nicotine-exposed groups (P <0.001, one-way analysis of variance). All data were evaluated at 72 hpf. Only statistically significant pair-wise comparisons in post-hoc analysis are shown. *Statistically B significant (P<0.05, total n=150).
|
Nicotine-induced hair cell toxicity in neuromasts of zebrafish embryos (brn3c)
Nicotine-induced hair cell damage in four neuromasts of Tg (brn3c:EGFP) embryos was evaluated (Fig. 3). The average number of hair cells in four neuromasts significantly decreased as nicotine concentration increased (control, 100%; nicotine 5 μM, 77.3%; nicotine 10 μM, 74.1%; nicotine 20 μM, 66.8%; P<0.05, ANOVA). Fish exposed to 40 μM nicotine were almost all dead at 120 hpf, therefore comparison with other groups was not possible. As shown in Fig. 4, significantly more TUNEL-positive cells (P<0.001, ANOVA) were observed in Tg (brn3c:EGFP) embryos as nicotine concentration increased.
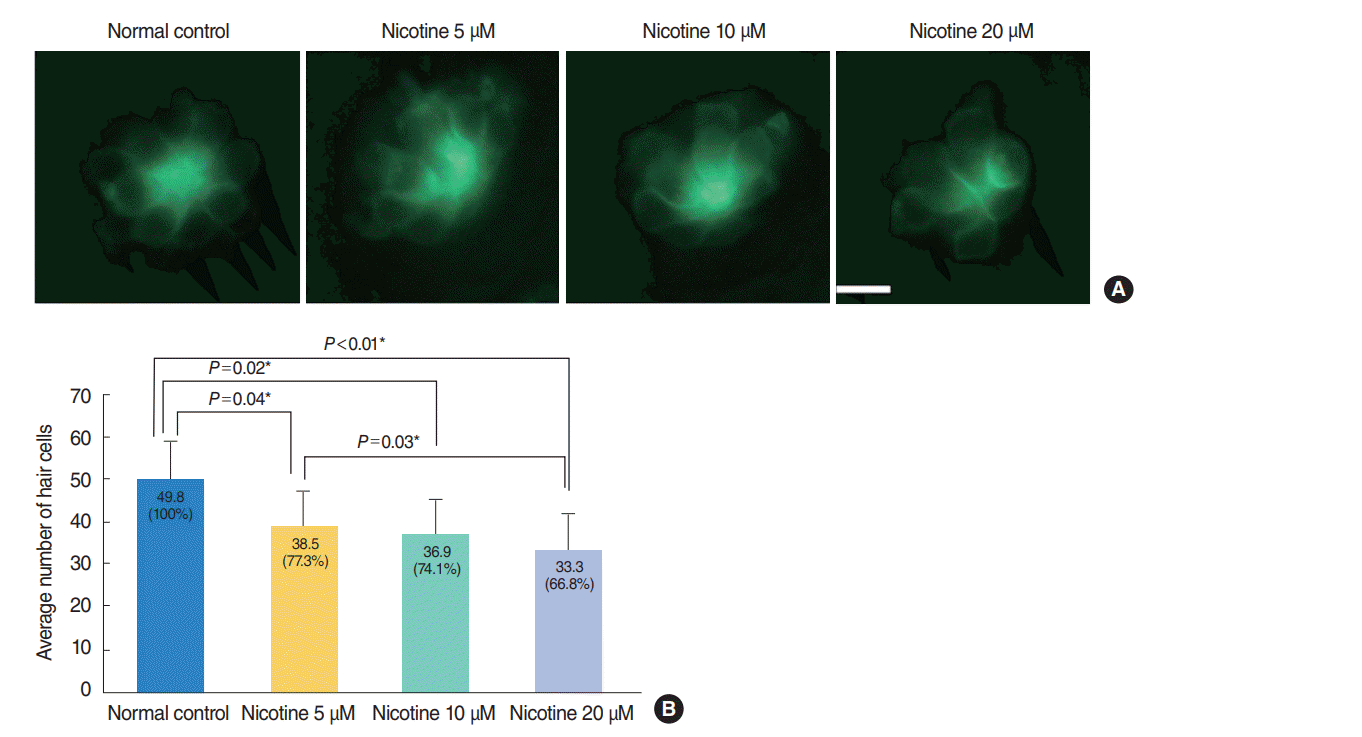 | Fig. 3.Nicotine-induced hair cell toxicity at 120 hours post-fertilization (hpf). The total number of hair cells was measured in four neuromasts (supraorbital [SO1 and SO2], otic [O1], and occipital [OC1]; total n=150). (A) The number of hair cells decreased in nicotine-exposed groups as shown by fluorescent microscopy (OC1, ×40). Scale bar=10 μm (×10). (B) Animals in the normal control group had an average of 49.8 hair cells. Nicotine exposure significantly decreased the number of total hair cells of the four neuromasts compared with that of the normal control, (P<0.001, one-way analysis of variance). At a concentration of 40 μM nicotine almost all the fish were dead, therefore statistical analysis was unavailable. All data were evaluated using transgenic (brn3c:EGFP) zebrafish at 120 hpf. Only statistically significant pair-wise comparisons in posthoc analysis are shown. *Statistically significant (P<0.05).
|
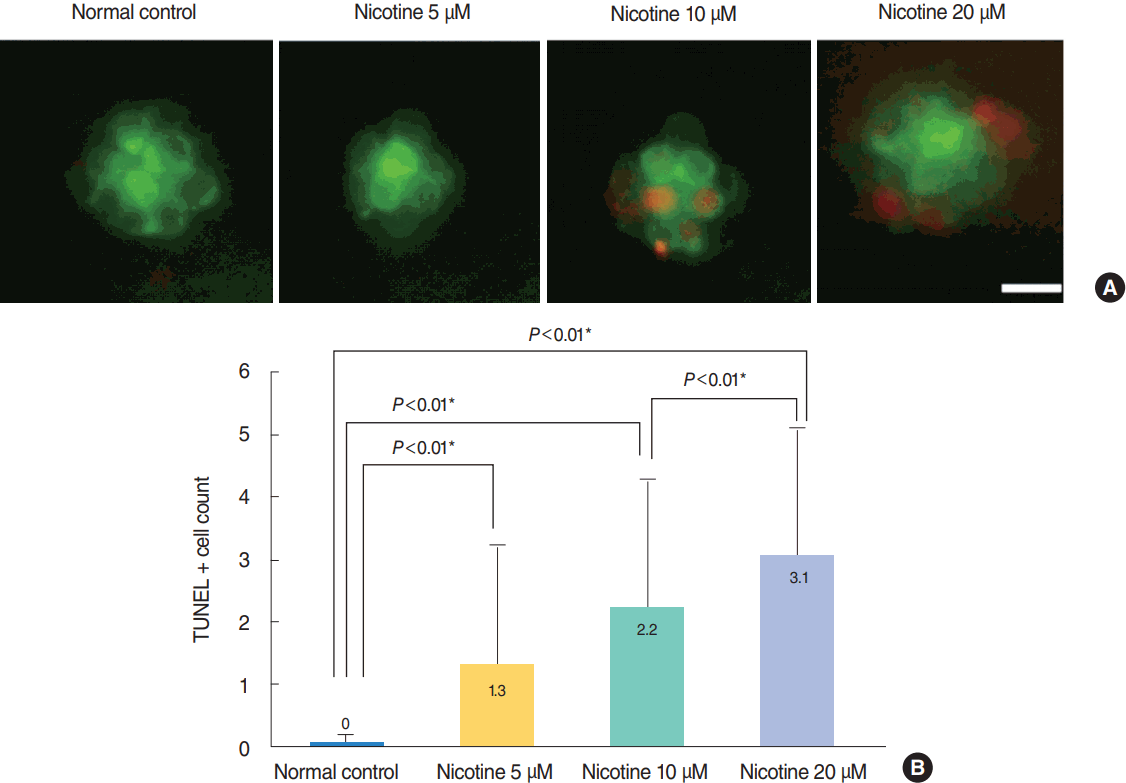 | Fig. 4.Nicotine-induced apoptosis in hair cells within neuromasts. Nicotine-induced apoptosis was confirmed by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay. (A) Green dots represent hair cells. Apoptotic cells appear as light red dots in the merged image obtained by fluorescent microscopy after the TUNEL reaction. All images were obtained using transgenic (brn3c:EGFP) zebrafish at 120 hours post-fertilization. Scale bar=10 μm (×40). (B) The average number of TUNEL-positive cells was measured in each neuromast (supraorbital [SO1, SO2], otic [O1], and occipital [OC1]; total n=50). At a concentration of 40 μM, the fish were almost all dead and statistical analysis was unavailable. The number of TUNEL-positive cells was significantly increased in nicotine-exposed groups (P<0.001, one-way analysis of variance). Only statistically significant pair-wise comparisons in post-hoc analysis are shown. *Statistically significant (P<0.05).
|
DASPEI staining in wild-type embryos
The average DASPEI-stained area in four neuromasts was analyzed among all experimental groups and the control group (n=6 for each concentration). Treatment of wild-type zebrafish with nicotine significantly increased the amount of cellular damage, as shown by a decrease in the average DASPEI-stained area expressed in hair cells (n=30, P<0.001, ANOVA) (Fig. 5). The average DASPEI area in four neuromasts was 384.3±113.9 μm2 in the control group, compared with 277.5±101.1 μm2 for fish treated with 20 μM nicotine. As in the TUNEL assay, fish treated with 40 μM nicotine were almost all dead after 120 hpf.
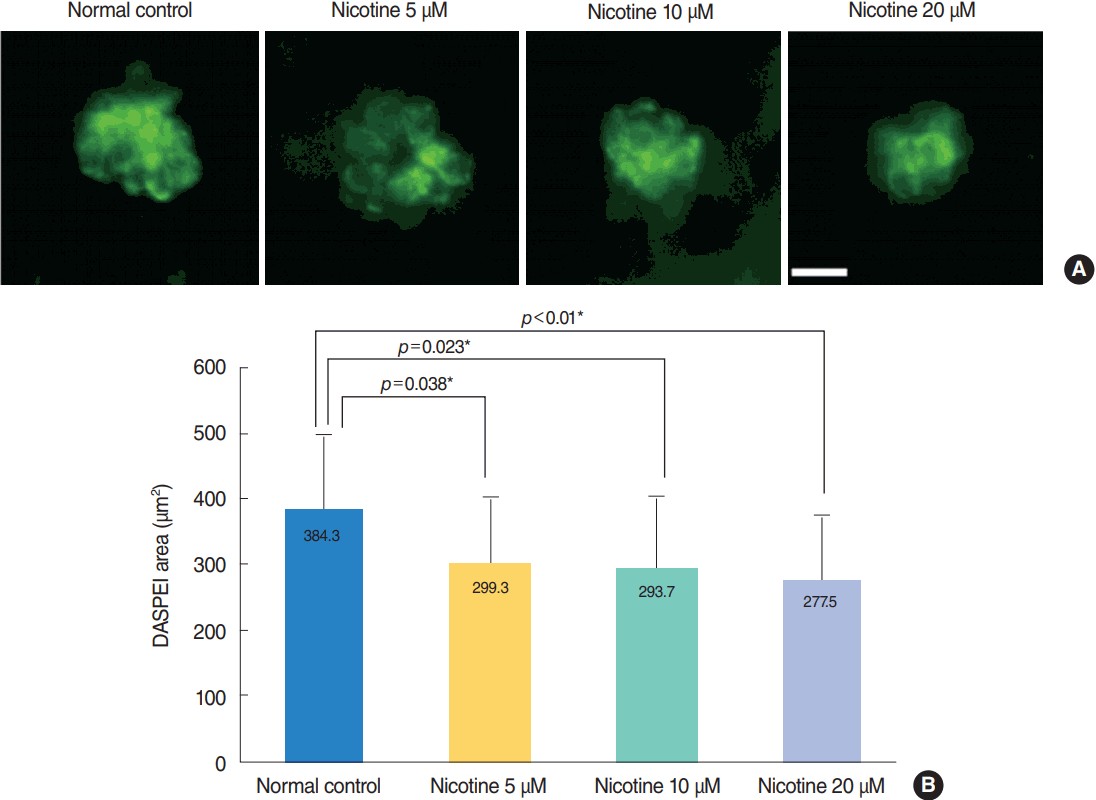 | Fig. 5.Analysis of hair cell damage by 2-(4-[dimethylamino]styryl)-N-ethylpyridinium iodide (DASPEI) assay. (A) Staining of hair cells with DASPEI showed that treatment with nicotine decreased the number of hair cells in neuromasts. Scale bar=10 μm (×40). (B) Average DASPEIstained area in four neuromasts (supraorbital [SO1, SO2], otic [O1], and occipital [OC1]; total n=30) significantly decreased as nicotine concentration increased (P<0.001, one-way analysis of variance). Fish exposed to 40 μM nicotine were almost all dead after 120 hours post-fertilization. Only statistically significant pair-wise comparisons in post-hoc analysis are shown. *Statistically significant (P<0.05).
|
Measurement of intracellular ROS generation in wild-type larvae
Representative result of ROS experiment is shown in Fig. 6. Positive control (125 μM neomycin) showed increased ROS in hair cells using DCHF-DA staining. In 5 μM nicotine, ROS generation was not different when compared with normal control, but was definitely more expressed in 40 μM nicotine.
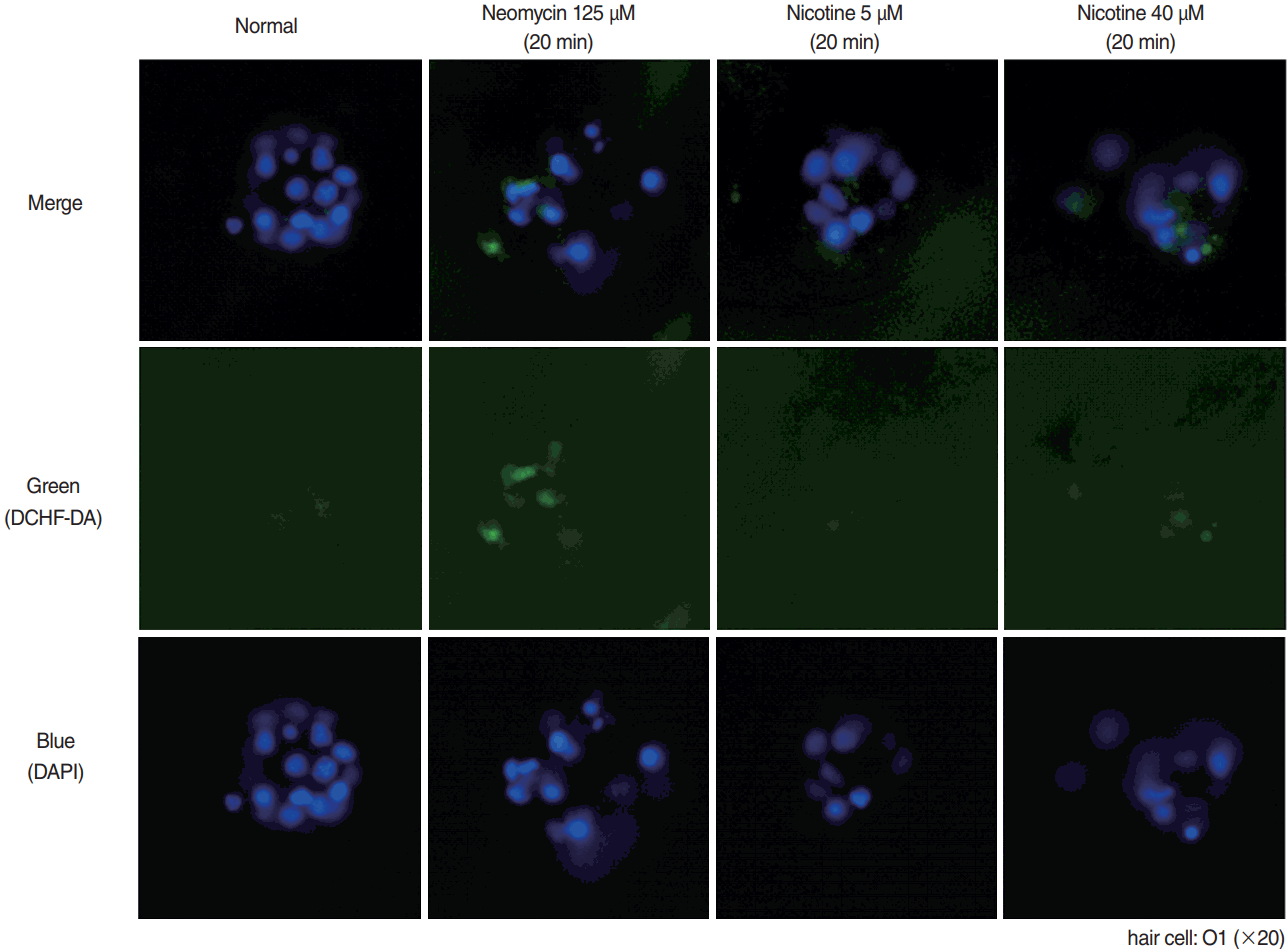 | Fig. 6.Reactive oxygen species (ROS) production by 2,7-dichlorodihydrofluorescein diacetate (DCHF-DA) assay. For evaluation of ROS generation, wild type zebrafish was used with DCHF-DA assay on hair cells as in green dots in the middle row. A 4´,6-diamidino-2-phenylindole (DAPI)-positive cells were observed as blue fluorescent dots as in the lower row. By merging those figures, the ROS production cells could be clearly identified as in the upper row. Neomycin as positive control showed strongest ROS generation. Despite not strong as in neomycin, there was increased ROS generation in hair cells in nicotine 40 μM. O1, otic.
|
Nicotine-induced ultrastructural changes by SEM
When observed with SEM, the kinocilia and the stereocilia bundles of hair cells were intact in the control group; however, the kinocilia of hair cells were destroyed in the 40 μM nicotine group (Fig. 7) and the 5 μM nicotine group showed fewer stereocilia bundles than the control group.
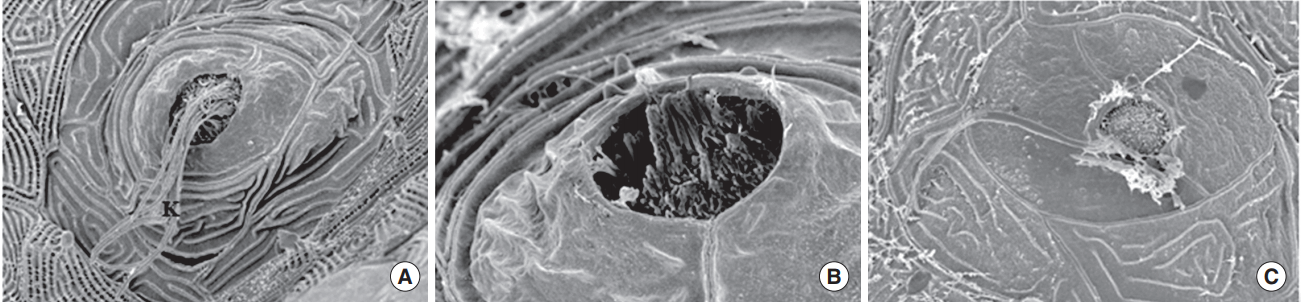 | Fig. 7.Scanning electron microscopy. When transgenic (brn3c:EGFP) zebrafish were exposed to nicotine for 120 hours, the kinocilia of hair cells in neuromasts were clearly observed in the normal control (A) and 5 μM nicotine group (B), but not in the 20 μM nicotine group (C). All images were obtained in three zebrafish at 120 hours post-fertilization for each group. Scale bar (at the bottom of each figure, one space)=5 or 10 µm.
|
Go to :
DISCUSSION
Nicotine is known to act through the activation of nAChRs, which can induce cellular apoptosis [21]. Previous studies reported that hair cells have nAChRs that are activated by the neurotransmitter acetylcholine, and that these nAChRs are crucial for establishment of innervation to the hair cells prior to hearing [18].
nAChRs are reported to be expressed in zebrafish embryos, and acetylcholinesterase-positive cells have been known to be essential for normal neuromuscular and neuronal development [5,22]. Because nicotine induces alterations in embryonic morphology, the zebrafish model has been used in various studies investigating the molecular and biological effects of nicotine exposure [5-7]. Several zebrafish nACHRs subunits are characterized in early development, and they have a high degree of sequence identity to nACHRs expressed in mammalian species [23]. The effects of prenatal nicotine exposure have been widely studied using zebrafish, and the zebrafish model is also used in various research studies investigating nicotine addiction, cognitive function, anxiolytic effects, and nAChR characterization [5,24].
In the present work, nicotine administration induced hair cell toxicity in the lateral line and embryotoxicity of zebrafish during embryonic development. Our results for TUNEL staining and DASPEI staining showed significantly increased apoptosis of hair cells as nicotine concentration increased. In the current study, the exposure of nicotine during zebrafish development seemed to increase the risk of embryotoxicity, caused significantly higher rates of abnormal morphology. However, hatching and mortality rates were higher in nicotine exposure groups than control group, but there was no significant dose-dependent trend. Nicotine is known to paralyze developing zebrafish when exposures occur during embryogenesis, and this leads to unhatching (paralyzed) or abnormal morphology (partially paralyzed) [25]. Our results partially correspond with previous studies that reported an increase in teratogenicity in a concentration-dependent manner following nicotine treatment [3,8]. Since in-well mortalities often cause increased toxicity to adjacent embryos, so this may have affected the increasing mortality as the nicotine concentration increase. However, there is a significant dose-dependent trend according to the nicotine concentration suggesting the teratogenic effect of nicotine.
Although heart rates were significantly lower in nicotine-exposed groups than the control group in our study, heart size was not different among groups and heart malformation was not observed in any of the groups. Our results for cardiac toxicity correspond with the findings of previous studies, which showed no heart defects in isolated nicotine exposure [9]. In contrast, when exposed to cigarettes, severe heart malformation including pericardial edema, reduced heart function was found in zebrafish [8,9]. Many chemicals in tobacco, including unknown and known ingredients such as polycyclic aromatic hydrocarbons are considered to cause cardiac toxicity in zebrafish [9]. Significant decrease in the heart rate of nicotine exposed zebrafish may have influenced on the development of the hair cells.
Nicotine is known to induce intracellular ROS generation in several kinds of cell lines and also in zebrafish [26,27]. Also in lateral line neuromasts of zebrafish, nicotine application provoked intracellular ROS generation which was demonstrated by our experiment. This ROS generation may have resulted in apoptosis of hair cells in neuromasts, which can be a possible mechanism of ototoxicity by nicotine.
Relatively few studies have investigated whether nicotine affects hearing in humans. In human population-based studies, current smoking [11], passive smoking [28] or past smoking [29] were all reported to be associated with hearing impairment after controlling for potential confounders. In addition, Durante et al. [15] reported that maternal tobacco exposure induced a significant decrease in transient evoked otoacoustic emissions amplitudes, suggesting an impact on outer hair cells. In a study of adolescents, maternal prenatal smoking exposure [30] or childhood nicotine exposure [14] were associated with higher pure-tone thresholds or abnormal transient evoked otoacoustic emissions amplitudes, respectively. Furthermore, few animal studies on this topic have been reported. In a study using rats, neonatal nicotine exposure reduced gap-induced prepulse inhibition suggesting damage of the normal development of auditory temporal processing by causing changes in cholinergic systems [16]. In the study of Abdel-Hafez et al. [18], nicotine caused structural damage of sensory hair cells, chiefly at the basal cochlear turn and outer hair cells, in adult guinea pigs. Especially, changes in stereocilia such as becoming bent and/or disorganized with a loss of their tip and side links were reported. Our SEM findings, which showed damage of kinocilia and the stereocilia bundles, correspond with those of Abdel-Hafez et al. [18]. Such damage of the hair cells and their stereocilia would result in clinical hearing loss, as recorded in humans [2,11].
Although several types of nAChRs have been described in zebrafish, the metabolism and half-life of nicotine in zebrafish are unknown [22]. We performed a literature review and pilot study to investigate the appropriate nicotine concentrations for the main research, and based on these findings used 5, 10, 20, 40 μM nicotine in the current study [3,5,22]. The mortality rate at 72 hpf in the 40 μM group was 9%, compared with 19% for the 20 μM group. However, when observed at 120 hfp, almost all of the fish in the 40 μM nicotine group were dead (>90% vs. 50% in 20 μM group) and further statistical analysis for the 40 μM group was not possible. This may be due to the high teratogenicity rate (44% for 40 μM nicotine at 72 hpf) but was very low survival when compared with the study of Parker and Connaughton [3], which showed a survival rate of approximately 40% in the 40 μM group at 5 days post-fertilization. Although the mortality rate at specific times should be compared between groups, surviving fish with abnormal morphology can also die at a later time therefore continuous monitoring is also very important.
We evaluated the hair cells in neuromasts at 120 hpf after 120 hours of nicotine exposure. According to our previous unreported experiment in normal zebrafish, neuromasts at 72 hpf (average of 6.8 hair cells per neuromast) and 96 hpf (average of 8.8 hair cells per neuromast) are not fully developed and show fewer hair cells. At about 120 hpf, hair cell numbers reach normal range (average of 12.5 hair cells per neuromast) of hair cells in each neuromasts.
In conclusion, according to our knowledge, our findings are the first study to investigate nicotine-induced hair cell toxicity in the neuromasts of zebrafish. The current results suggest that nicotine induces dose-dependent mortality, teratogenicity, and hair cell toxicity through the induction of apoptosis and damage of stereocilia and kinocilia.
Go to :
 XML Download
XML Download