

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Diabetes mellitus (DM) is one of metabolic diseases in which there are high blood sugar levels over a prolonged period, and if left untreated, diabetes can cause many complications, which include cardiovascular disease, chronic renal disease, neuropathy, and retinopathy [1]. Furthermore, inflammatory airway disease appears with more frequent relapse and longer duration of symptoms in the patients with DM [2,3].
High glucose (HG) is an essential prerequisite to pathogenesis of the complications of DM, and HG is well known to induce mitochondrial superoxide production resulting in systemic inflammatory response via various signaling pathways [1]. In particular, the mechanisms of tissue damaging by HG have been well studied in retinal capillary endothelial cells, mesangial cells in the renal glomerulus, neurons of peripheral nerves, skin keratinocytes, and cancer cells [1,4-8]. In respiratory system, several studies have demonstrated that HG can lead to structural changes in lungs, such as fibrosis, edema, and dilatation or destruction of alveoli, resulting in declined lung function [9,10] and the aggravation of respiratory disease by promoting local inflammation, which may promote pulmonary infection [2,11]. However, there are few studies examined the effect of HG on airway epithelial cells [12]. The effects of the production of mucus in human airway epithelial cells and the mechanisms involved are not clear to date.
Therefore, the aims of this study were to investigate the effects of HG on the expressions of major secretary mucins and to identify the signaling pathway involved in human airway epithelial cells.
Go to : 
MATERIALS AND METHODS
Cell culture and treatment
Mucin-producing human NCI-H292 airway epithelial cells were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS; Hyclone Laboratories, South Logan, UT, USA). The cells were grown at 37°C in 5% CO2 fully humidified air and were subcultured twice weekly. The cells were seeded in wells of a 6-well plate at 1×105 cells/well. When growth was confluent, the cells were incubated in RPMI 1640 medium containing 0.5% FBS for 24 hours. The cells were then rinsed with serum-free RPMI 1640 medium and exposed to certain concentrations of D-Glucose (Sigma-Aldrich, St. Louis, MO, USA) for the indicated times. The cell proliferation and viability of NCI-H292 cells was quantified by EZ-Cytox Cell Viability Assay Kit (Daeil Lab Services, Seoul, Korea). And also the changes of cell morphology was checked by using microscope in this study. This study was approved by the Institutional Review Board for human studies at the Yeungnam University Medical Center (YUH-13-0375-033).
To investigate the signaling pathway of HG-induced MUC5B expression, U0126 (EMD Chemicals, Gibbstown, NJ, USA), SB203580 (Biomol Research Laboratories, Plymouth Meeting, PA, USA), and AG1478 (Sigma-Aldrich) as specific inhibitors, were used to pretreat the cells for 1 hour before exposure to the indicated concentrations of glucose. For the controls, the cells were incubated with 5 mM of glucose, which is the physiologic concentration in human body [7,8], for the same amount of time.
RT-PCR analysis of MUC2, MUC5AC, and MUC5B mRNA expression
Isolation of total RNA from the cultured cells was performed according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA, USA). Each sample was reverse transcribed into cDNA using the GeneAmp RNA PCR Core Kit (Applied Biosystems). The primer sequences used in the PCR were 5´-CAG CAC CGA TTG CTG AGT TG-3´ (sense) and 5´-GCT GGT CAT CTC AAT GGC AG-3´ (antisense) for MUC2 (140 bps), 5´-TCC ACC ATA TAC CGC CAC AGA-3´ (sense) and 5´-TGG ACG GAC AGT CAC TGT CAA C-3´ (antisense) for MUC5AC (103 bps), and 5´-CAC ATC CAC CCT TCC AAC-3´ (sense) and 5´-GGC TCA TTG TCG TCT CTG-3´ (antisense) for MUC5B (245 bps) [13,14]. RNA integrity and the success of the reverse transcription (RT) reaction were monitored by PCR amplification of the transcripts for the GAPDH gene [13]. The PCR products were electrophoresed on a 2% agarose gel, followed by staining with ethidium bromide, and visualization by ultraviolet fluorescence. Semiquantitative analysis of the reverse transcriptase-polymerase chain reaction (RT-PCR) product was performed on the scanned gel images, and the intensity of the PCR product was measured using commercially available imaging software (Scion, Frederick, MD, USA). The relative intensity of the individual bands on the gel images were determined as the ratio of the intensities of MUC2, MUC5AC, and MUC5B to the intensity of GAPDH.
Real-time PCR analysis of MUC5B and EGFR mRNA expression
Real-time PCR was performed with the LightCycler Fast Start DNA Master SYBR Green kit (Roche Applied Science, Mannheim, Germany) using 0.5 μL of cDNA, corresponding to 25 ng of total RNA in a 10-μL final volume, 2.5 mM MgCl2 and 0.5 μM of each primer (final concentration). Quantitative PCR was performed using a LightCycler (Roche Applied Science) for 45 cycles at 95°C for 10 seconds, specific annealing temperature for 5 seconds and 62°C for 10 seconds. Data were normalized versus GAPDH. Amplification specificity was evaluated using melting curve, following the manufacturer’s instructions (Roche Applied Science).
Enzyme-linked immunosorbent assay analysis of MUC5B protein
The protein levels of MUC2, MUC5AC, and MUC5B were determined by enzyme-linked immunosorbent assay (ELISA). Samples of cell supernatants and cell lysates were prepared in phosphate-buffered saline at several dilutions, and incubated at 40°C in a 96-well plate until dry. The plates were then washed three times with phosphate buffered saline (PBS), blocked with 2% bovine serum albumin for 1 hour at room temperature, rewashed three times with PBS, and incubated with primary antibody for MUC2, MUC5AC or MUC5B (diluted at 1:200 with PBS containing 0.05% Tween 20) for 1 hour. Wells were then washed three times with PBS, and horseradish peroxidase (HRP)-conjugated secondary antibody for MUC2, MUC5AC or MUC5B was added to wells, and 4 hours later, color was developed using 3,3´,5,5´-tetramethylbenzidine peroxidase solution and stopped with 2N-H2SO4. Optical densities were measured using an ELISA reader (EL800, BioTek Instruments, Winooski, VT, USA) at 450 nm. The results are expressed as percentages of baseline controls.
Western blot analysis of EGFR, ERK1/2 mitogen-activated protein kinase (MAPK), and p38 MAPK phosphorylation
Mucin-producing human NCI-H292 airway epithelial cells were seeded in wells of a 6-well plate and treated with glucose for the indicated times and concentrations. The cells were then washed with cold PBS, exposed to trypsin, and formed into pellets at 700×g for 10 minutes at 4°C. The pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail). The preparation was clarified by centrifugation, and the supernatant was saved as a whole-cell lysate. The proteins (50 μg) were separated using 10% reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electroblotted in 20% methanol, 25 mM Tris, and 192 mM glycine onto a nitrocellulose membrane, which was then blocked with 5% nonfat dry milk in 25 mM Tris-HCl, 150 mM NaCl, and 0.2% Tween 20, and then incubated with the indicated primary antibody of EGFR (Cell Signaling Technologies, Boston, MA, USA), ERK1/2 (Cell Signaling Technologies), or p38 (Santa Cruz Biotechnology, Dallas, TX, USA) for 4 hours. Subsequently, the membrane was washed and incubated for 1 hour with secondary antibody of EGFR (Cell Signaling Technologies), ERK1/2 (Cell Signaling Technologies), or p38 (Cell Signaling Technologies) conjugated to HRP, rewashed, and developed using an enhanced chemiluminescence reagent kit (Perkin Elmer Life Sciences, Boston, MA, USA) and exposed to X-ray film for 10 seconds.
Cell transfection with siRNA for p38 MAPK
Predesigned siRNA targeting p38 MAPK and control siRNA for p38 MAPK were purchased from Invitrogen. The sequences of p38 siRNA were 5´-AUG AAU GAU GGA CUG AAA UGG UCU G-3´ (sense) and 5´-CAG ACC AUU UCA GUC CAU CAU UCA U-3´ (antisense). The transfection efficiency of p38 MAPK siRNA was verified to be over 90% in the mucin-producing human NCI-H292 airway epithelial cells. Transfection was performed according to the manufacturer’s instruction (Invitrogen). Briefly, human NCI-H292 airway epithelial cells were seeded in wells of a 6-well plate at 1×105 cells/well and incubated in RPMI 1640 medium. When the cells were confluent, OPTI-MEM I Reduced Serum Medium (Invitrogen) was added. Then, p38 MAPK siRNA and Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) were incubated together in OPTI-MEM I Reduced Serum Medium to form a p38 MAPK siRNA-Lipofectamine complex. This complex-containing medium was then added to each well containing the cells to a final p38 MAPK siRNA concentration of 20 mM. After 24 hours of transfection with p38 MAPK siRNA, the cells were exposed to the indicated concentrations of glucose and then harvested for RT-PCR analysis of MUC5B mRNA expression. The same procedure was performed with control siRNA.
Statistical analysis
Statistical analysis was performed using SPSS ver. 12.0 (SPSS Inc., Chicago, IL, USA). The mean for each of the obtained quantitative values was calculated. Comparisons were made using the Student t-test, analysis of variance, and Mann-Whitney U test. For all tests, P-value less than 0.05 was considered statistically significant.
Go to :
RESULTS
Effect of HG on the expression of major secretary airway mucin genes
To investigate the effect of HG on MUC2, MUC5AC, and MUC5B expression in NCI-H292 cells, the cells were incubated with different concentrations of glucose for 8 hours. Results of RT-PCR showed that glucose significantly induced MUC5B mRNA expression at concentrations higher than 15 mM, compared with the control group (5 mM). However, glucose did not induce MUC2 and MUC5AC mRNA expression at any concentrations (Fig. 1). Real-time PCR analysis and ELISA were performed to investigate the effect of HG on MUC2, MUC5AC, and MUC5B expression with dose- and time-dependent experiments. NCI-H292 cells were treated with different concentrations of glucose for 8 hours or 20 mM of glucose for different times. As the results obtained in RT-PCR, MUC5B expression was significantly increased by glucose at concentrations higher than 15 mM, compared with the control group (5 mM). However, glucose did not induced MUC2 and MUC5AC expression (Fig. 2A, B). And in time-dependent experiment, MUC5B mRNA expression was significantly increased at all times after exposure of glucose (20 mM). However, MUC2 and MUC5AC mRNA expression were not induced by glucose (Fig. 2C).
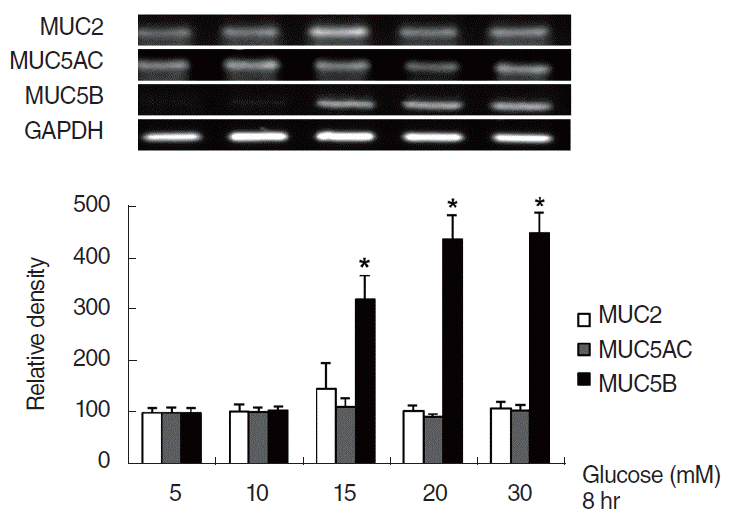 | Fig. 1.Effect of high glucose (HG) on the expression of secretary airway mucins in NCI-H292 cells. Reverse transcriptase-polymerase chain reaction showed that HG (>15 mM) significantly induced MUC5B mRNA expression. However, glucose did not induce MUC2 or MUC5AC mRNA expression at any concentration examined. Images are representative of three separate experiments performed in triplicate. Bars indicate the mean±standard deviation of three independent experiments performed in triplicate. *P<0.05.
|
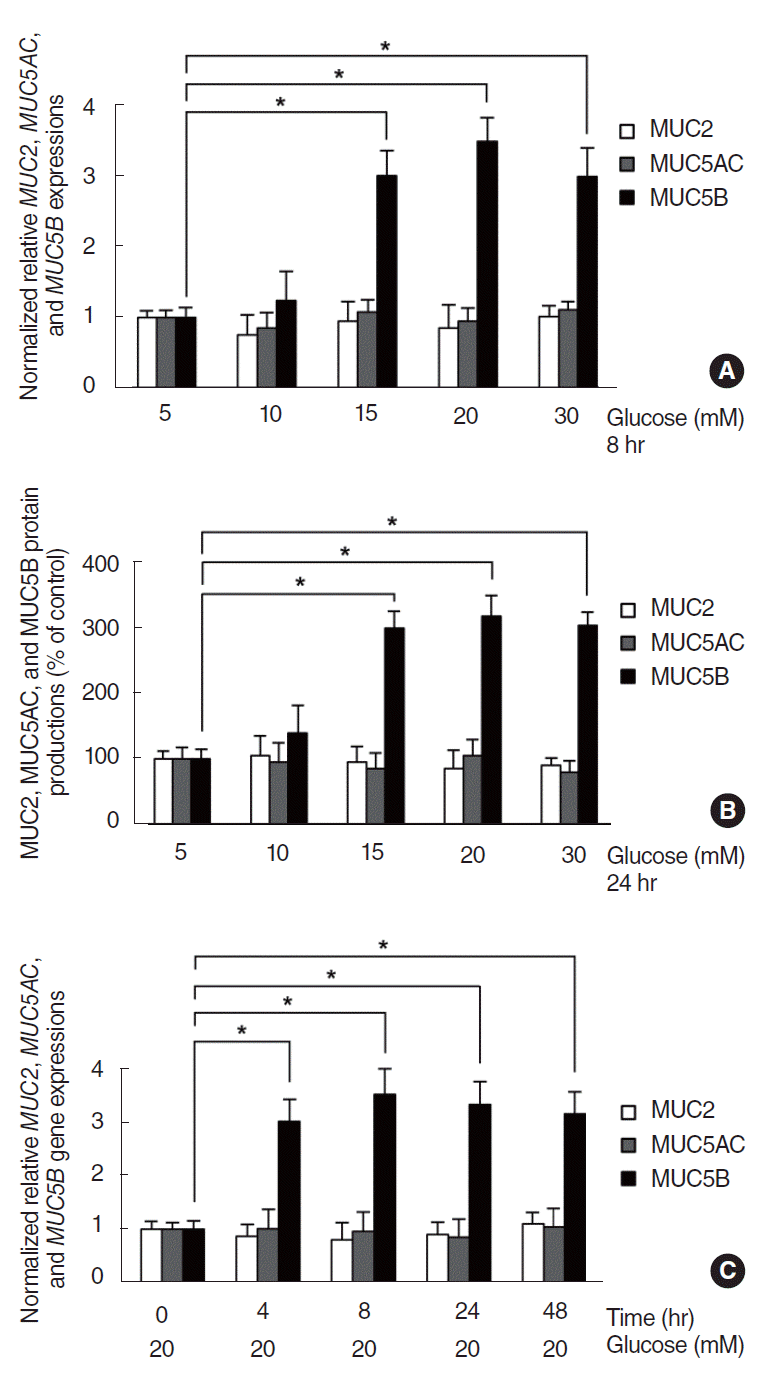 | Fig. 2.Effect of high glucose (HG) on MUC5B expression in NCI-H292 cells. Real-time polymerase chain reaction (PCR) and enzyme-linked immunosorbent assay (ELISA) showed that MUC5B expression was significantly increased at concentrations higher than 15 mM, compared with the control group (5 mM). However, glucose did not induced MUC2 and MUC5AC expression (A, B). Real-time PCR showed that MUC5B mRNA expression was significantly increased at all times after exposure of glucose (20 mM) (C). Images are representative of three separate experiments performed in triplicate. Bars indicate the mean±standard deviation of three independent experiments performed in triplicate. *P<0.05.
|
Effect of HG on the activation of EGFR
To investigate the effect of HG on EGFR activation, NCI-H292 cells were stimulated with different concentrations of glucose (5, 10, 15, 20, or 30 mM) for 8 hours, and then EGFR mRNA expression and phosphorylation were examined. Real-time PCR showed that EGFR mRNA expression was significantly increased at concentrations higher than 15 mM and peaked at 20 mM of glucose, compared with the control group (5 mM) (Fig. 3A). Western blot showed that HG (20 mM) significantly activated the phosphorylation of EGFR with the lapse of time (Fig. 3B).
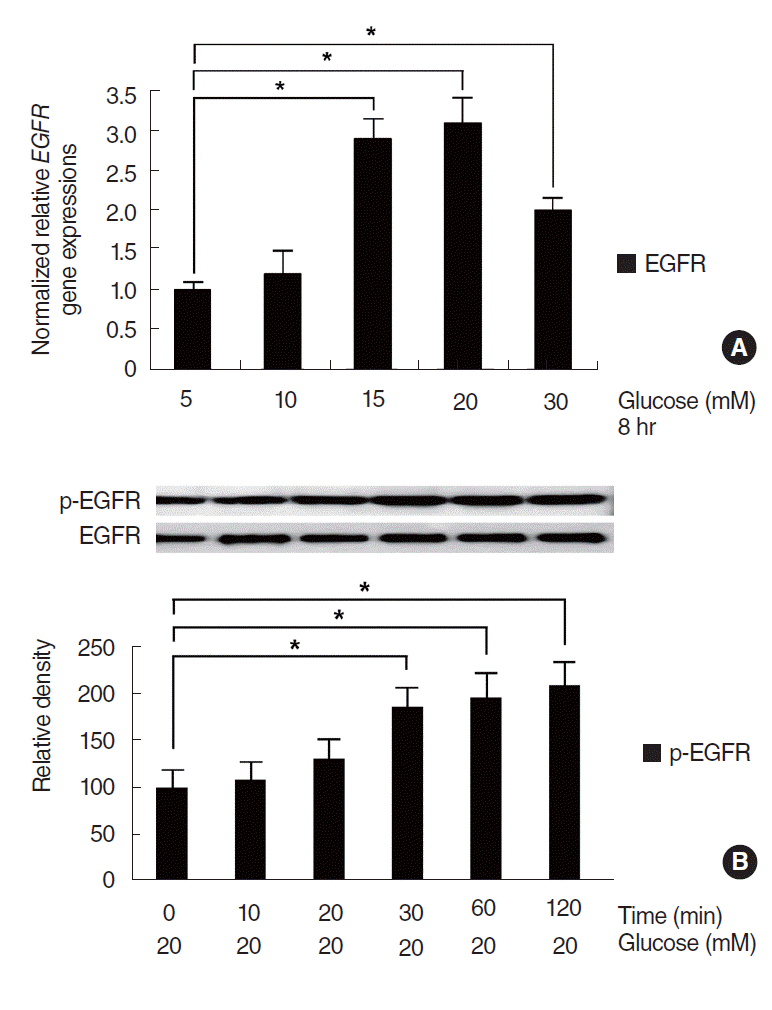 | Fig. 3.Effect of high glucose (HG) on the activation of epidermal growth factor receptor (EGFR) in NCI-H292 cells. Real-time polymerase chain reaction showed that EGFR mRNA expression was significantly increased at concentrations higher than 15 mM and peaked at 20 mM of glucose, compared with the control group (5 mM) (A). Western blot showed that HG significantly activated the phosphorylation of EGFR with the lapse of time (B). Images are representative of three separate experiments performed in triplicate. Bars indicate the mean±standard deviation of three independent experiments performed in triplicate. *P<0.05.
|
The roles of EGFR, ERK1/2 MAPK, and p38 MAPK on HG-induced MUC5B expression
The phosphorylation of ERK1/2 and p38 MAPK were examined to evaluate the intracellular mechanisms of HG-induced MUC5B expression. Western blot showed that HG increased the phosphorylation of p38 MAPK, but not that of ERK1/2 MAPK (Fig. 4A). To investigate the involvements of the EGFR, ERK1/2 MAPK, and p38 MAPK signaling pathway on HG-induced MUC5B expression, the cells were stimulated with AG1478 as a specific inhibitor of EGFR, U0126 as a specific inhibitor of ERK1/2 MAPK, and SB203580 as a specific inhibitor of p38 MAPK for 1 hour before being exposed to glucose (20 mM). RT-PCR and ELISA showed that SB203580 and AG1478 significantly attenuated HG-induced MUC5B expression, but that U0126 had no effect (Fig. 4B, C). In addition, to evaluate the correlation between the EGFR and p38 MAPK signaling pathways, we examined the phosphorylation of p38 MAPK after pretreating cells with AG1478 for 1 hour before treatment with glucose (20 mM) in NCI-H292 cells. Western blot showed that AG1478 significantly attenuated HG-induced the phosphorylation of p38 MAPK (Fig. 4D).
 | Fig. 4.Roles of epidermal growth factor receptor (EGFR), ERK1/2 mitogen-activated protein kinase (MAPK), and p38 MAPK on high glucose (HG)-induced MUC5B expression. (A) Western blot showed that HG significantly activated the phosphorylation of p38 MAPK with the lapse of time, but that HG did not activate the phosphorylation of ERK1/2 MAPK. (B, C) Reverse transcriptase-polymerase chain reaction (RT-PCR) and enzyme-linked immunosorbent assay (ELISA) showed that SB203580 (a p38 MAPK inhibitor) and AG1478 (an EGFR inhibitor) significantly attenuated HG-induced MUC5B expression, whereas U0126 (an ERK1/2 inhibitor) had no effect. (D) Western blot showed that AG1478 significantly attenuated the HG-induced phosphorylation of p38 MAPK. (E and F) RT-PCR and ELISA showed that knockdown of p38 MAPK by p38 MAPK siRNA significantly blocked HG-induced MUC5B expression. Images are representative of three separate experiments performed in triplicate. Bars indicate the mean±standard deviation of three independent experiments performed in triplicate. *P<0.05.
|
Go to :
DISCUSSION
The effect of HG on the expressions of major secretary airway mucin genes, such as, MUC2, MUC5AC, and MUC5B were checked in this study. In results, only MUC5B expression was significantly increased by HG exposure. Therefore, the present study was conducted to confirm the signaling pathway responsible for HG-induced MUC5B expression in human airway epithelial cells.
The prevalence of DM is increasing worldwide due to aging, sedentary life styles, and growth of obesity population. Consequently, diabetic complications are also increasing. In particular, inflammatory airway disease appears with more frequent relapse and longer duration of symptoms in patients with diabetes. And response to treatment is not good for it. The prevention and treatment of complications related with airway inflammation are considered to be a most important for general care of DM patients. Glucose toxicity is the main cause of DM complications and causes damage to many types of tissues via multiple signaling pathways: the polyol pathway activation, intracellular production of advanced glycation end products, the protein kinase C (PKC) activation, the hexosamine pathway activation, and the EGFR activation [1,4-9]. Recently, several studies have demonstrated that HG causes structural and functional changes in respiratory system [9,10] and promotes the aggravation of respiratory disease by promoting local inflammation and infection [2,11]. However, the mechanisms related to worsening of inflammatory airway diseases caused by HG were not well understood.
EGFR is a 170-kDa cell surface receptor with intrinsic tyrosine kinase activity. EGFR is activated via two mechanisms. First is the EGFR activation by distinct ligands such as epidermal growth factor (EGF). And second is the EGFR autophosphorylation by interaction with numerous intermediary effector molecules, such as, G protein-coupled receptors, cytokines, hormones or cellular stress [15]. Recently, some studies were demonstrated that HG activates the EGFR in a nonligand-dependent manner in different types of cells. In mesangial cells, HG induces PKCβ1-Akt activation and collagen I upregulation via EGFR-phospholipase Cγ1 signaling [5]. Furthermore, HG enhances interleukin 8 (IL-8) production via EGFR-ERK pathway in keratocytes [7], and promotes cell proliferation via the induction of EGF expression and transactivation of EGFR in pancreatic cancer cell [8]. However, the effect of HG on EGFR activation in human airway epithelial cells has not been previously examined. The results of this study showed that HG significantly increased EGFR mRNA expression and phosphorylation in human airway epithelial cells.
Airway epithelial surface signaling through EGFR is a convergent pathway producing innate immune responses to multiple environmental stimuli [16]. Many studies have suggested that the EGFR activation leads to airway mucin production [17-19], neutrophil recruitment [20], and airway epithelial remodeling [21]. In particular, it has been well known that the upregulation of mucin expression is associated with EGFR activation in inflammatory airway diseases [16-19]. The results of this study demonstrated that AG14078 (an inhibitor of EGFR) significantly attenuated HG-induced MUC5B mRNA expression, which means EGFR activation by HG contributes to MUC5B expression in human airway epithelial cells.
To date, about 20 types of mucin genes have been deduced. Mucins are highly glycosylated proteins responsible for the viscoelastic properties of mucus [22]. MUC5AC and MUC5B are representative of secreted type mucin genes in human airway [23,24]. These are abnormally upregulated in the inflammatory airway diseases, such as, chronic bronchitis, chronic obstructive pulmonary disease, asthma, and cystic fibrosis [22], and its expression is induced in response to a wide variety of stimuli, such as, nerve activation and inflammatory cytokines, including IL-1β, IL-6, IL-9, IL-13, and tumor necrosis factor-α [25,26]. In addition, several reports have demonstrated that the signaling pathways associated with EGFR or MAPK are responsible for MUC5B expression in airway epithelial cells, for example, Mycoplasma pneumonia was reported to induce MUC5B expression by activating the EGFR signaling pathway [17], and MUC5B expression was found to be up-regulated by PMA (phorbol-12-myristate-13-acetate) through the EGFR/MAPK-independent and -dependent pathways [18]. Therefore, this study focused on the EGFR-mediated ERK1/2 and p38 MAPK signaling pathway. The results of this study showed that HG significantly activated the phosphorylation of p38 MAPK via EGFR activation. However, the phosphorylation of ERK1/2 MAPK was not induced. In addition, the knockdown of p38 MAPK by siRNA significantly blocked HG-induced MUC5 expression. These findings suggested that the sequential activation of EGFR/p38 MAPK is involved in HG-induced MUC5B mRNA expression in human airway epithelial cells.
The events leading to EGFR activation by HG have not been determined. Rapid release of membrane-bound EGF ligands, such as, heparin-binding EGF and the generation of reactive oxygen species by HG are potential mechanism for EGFR activation by HG [27,28]. Although, in present study, the precise mechanism was not identified about the EGFR activation by HG, further studies are required. However, the findings of this study suggest for first time that HG causes EGFR activation in human airway epithelial cells, and that EGFR-mediated p38 MAPK activation was involved in HG-induced MUC5B expression.
The results of this study suggest that HG induces MUC5B expression via sequential activation of the EGFR/p38 MAPK signaling pathway in human airway epithelial cells and provide information supporting the notion that HG might affect the severity and duration of inflammatory airway diseases in DM patients.
Go to :
 XML Download
XML Download