

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Otitis media (OM) is a multifactorial disease caused by many agents, including viral and bacterial infections, biofilm formation, congenital anomaly, and environmental factors such as smoking, and air pollution [1]. Several epidemiologic studies conducted over the last decade have investigated the relationship between ambient air pollution and OM in human populations [2]. Children living in areas with high concentrations of particulate matter (PM) and SO2 have demonstrated significantly higher rates of OM than those living in the control areas with lower concentrations of PM [3]. Rovers et al. [4] suggested that the level of SO2 emissions is one of the important risk factors of OM in many industrialized Western countries. A large cohort study also revealed the close relationship between the prevalence of OM and air quality [5].
The invasion of Asian sand dust (ASD) is a seasonal phenomenon in Asia, which arises from the Gobi and Taklimakan desert plains of China. ASD is known to spread over large areas of East Asia every spring, including East China, North and South Korea, and Japan. ASD is composed of PM less than 10 μm in diameter (PM10), as well as several hazardous chemical compounds and microbial agents.
ASD is known to cause various human diseases, including allergies, chronic pulmonary injury, and cardiovascular and respiratory dysfunction [6,7]. Additionally, recent studies have demonstrated that ASD significantly aggravates respiratory inflammation and mucin production in murine lungs [6,8]. Therefore, the annual ASD event has become a serious human health concerns, especially with the increase of industrial pollutants in China.
The major aim of this study was to investigate the potential risk of ASD on the development of OM. To this end, we investigated the cytotoxic effect of ASD and the changes of mucin gene and inflammatory cytokine expression in human middle ear epithelial cells (HMEECs). Additionally, we evaluated the histopathologic changes in the middle ears of rats injected with ASD.
Go to : 
MATERIALS AND METHODS
Preparation of ASD
ASD was collected from the atmosphere on March 16, 2009 outside the Gachon University building in Korea, using a high volume air sampler (HV500F; Sibata Scientific Technology, Ltd., Saitama, Japan) at a flow rate of 500 L/min. These ASD samples were prefiltered into filter packs (Prefilter AP, 124 mm; EMD Millipore, Bedford, MA, USA) and sieved through a filter with a 10-μm pore size, after mixing with phosphate buffered saline (PBS) in a 15-mL tube. The filtered ASD particles were sterilized in an autoclave at 121°C for 15 minutes; subsequently, these particles were weighed. Each sample was transferred to a 1.5-mL tube. The filtered PM was stored at –20°C until further use. The particle diameter of the samples was measured (a total of 600 particles) under a scanning electron microscope (JSM-5800; JEOL Ltd., Tokyo, Japan). The mean distribution peak of the ASD particle diameter was observed at 6 μm. Chemical components of the ASD particles were analyzed via X-ray fluorescence spectrometry (PHILIPS pw2404; Philips, Eindhoven, The Netherlands) at the Korea Basic Science Institute. The chemical composition of ASD was determined to be as follows: 78.4% SiO2, 9.35% Al2O3, 2.52% K2O, 2.41% Na2O, 2.06% Fe2O3, and 1.74% CaO. MaO, TiO2, P2O5, and MnO made up less than 0.1% of the total composition.
Cell culture and reagents
HMEECs (kindly provided by Dr. David J. Lim, House Ear Institutes, Los Angeles, CA, USA), immortalized with the E6/E7 genes of human papilloma virus type [9] were maintained in a mixture of Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) and bronchial epithelial basal medium (BEBM) (Lonza, Walkersville, MD, USA) (1:1). The cells were maintained at subconfluence in a carbon dioxide-enriched (95% air, 5% CO2) humidified atmosphere, at 37°C. In order to study the effects of ASD, the cells were grown to 60% confluence in six-well culture plates at 37°C in a carbon dioxide-enriched (95% air, 5% CO2) humidified atmosphere. These cells were starved for 2 hours; these were exposed to ASD and subsequently incubated for 24 hours. The experimental group received various concentrations of ASD (50–400 μg/mL) while the control group did not receive ASD.
Cell viability assay
Cell viability was measured using the cell counting kit (CCK-8, Dojindo Laboratories, Kumamoto, Japan). HMEECs were seeded in 96-well plates, with each well containing 1×104 cells. The following day, cells were treated with 0, 6.25, 12.5, 25, 50, 100, 200, 300, or 400 μg/mL of ASD. CCK-8 solution was added to each well after 24 hours, and the plates incubated for 150 minutes at 37°C. The contents of the plates were mixed using a shaker (at room temperature for 5 minutes), and the optical density was measured at 450 nm using a microplate reader (Spectra Max plus 384; Molecular devices, Sunnyvale, CA, USA).
Real-time reverse transcriptase polymerase chain reaction
Real-time polymerase chain reaction (RT-PCR) was performed using a LightCycler 480 Real-Time PCR System (Hoffmann-LaRoche, Basel, Switzerland). Each reaction mixture contained 10 μL of LightCycler 480SYBR Green I Master (Hoffmann-LaRo che), 1 μL of 4 pmol sense and antisense primers, and 0.4 μL of cDNA, in a final volume of 20 μL. Reaction mixtures were incubated at 95°C for 5 minutes to activate the FastStart Taq DNA Polymerase; this was followed by amplification for 50 cycles (one cycle: 15 seconds at 95°C, 30 seconds at 60°C, and 30 seconds 72°C). The data was analyzed using the LightCycler 480 software 1.5 (Hoffmann-LaRoche). Target mRNA expression was normalized to that of GAPDH, and calculated using the comparative Ct method. The specific primers of genes used for realtime PCR are listed in Table 1.
Table 1.
Oligonucleotide primer sequences for quantitative reverse transcriptase polymerase chain reaction
![]()
Western blot analysis
HMEECs were stimulated with 0, 50, 100, 200, or 400 μg/mL of ASD for 24 hours. After treatment of HMEECs with ASD, the medium was removed and the cells were washed twice in PBS (10 mM, pH 7.4). Lysis buffer (0.2 mL, PRO-PREP; INtRON, Seongnam, Korea) was added, and the cells were incubated for 1 hour at –20°C. The cells were then centrifuged at 13,000 ×g for 10 minutes at 4°C. The supernatant containing the total cell lysate was collected and stored at –80°C. Protein concentration of the lysates was measured using Quick Start Bradford 1 dye reagent (Bio-Rad, Hercules, CA, USA). Equal quantities of protein were analyzed using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. The protein content of the gels was transferred to a polyvinyl difluoride membrane (Hybond P; Amersham Biosciences Corp., GE Healthcare, Buckinghamshire, UK), and the membranes blocked with 25 mM Tris and 120 mM NaCl and 0.05% Tween-20 containing 5% (w/v) skim milk for 1 hour at room temperature. Membranes were probed with antibodies against COX-2 (1:200; Santa Cruz Biotechnology, Dallas, Texas, USA) and β-Actin (1:3,000; Sigma-Aldrich, St. Louis, MO, USA), followed by peroxidase-conjugated antimouse IgG (1:5,000; Vector Laboratories, Burlingame, CA, USA). The membranes were developed using an ECL detection kit (Pierce, Rockford, IL, USA) and the signal captured using an image reader (LAS3000; Fuji Photo Film, Tokyo, Japan).
Animal maintenance and experimental design
Fifty pathogen-free, male Sprague Dawley rats weighting 150–200 g (Orient Bio, Seongnam, Korea) were used for this study. The protocols used in this study were approved by the Institutional Animal Care and Use Committee (IACUC); the animals were cared for according to the Guidelines of Animal Experiments of the Korea University Guro Hospital. The rats were assigned randomly to one of 5 groups: 4 groups received a unilateral tympanic membrane ASD injection (on days 1, 3, 5, and 14; each group, n=5) and the control group received a PBS vehicle injection (vehicle control group, n=5). ASD (400 μg/mL) dissolved in PBS was injected into all experimental mice (4 groups) at all time-points using a 27-gauge spinal needle. The rats were injected with ASD and subsequently sacrificed on days 1 (group 1), 3 (group 2), 5 (group 3), or 14 (group 4) postprocedure. Anesthesia was performed with an intraperitoneal injection of ketamine hydrochloride (30 mg/kg) and xylazine hydrochloride (5 mg/kg).
Histological evaluation
Five rats from each experimental group (groups 1, 2, 3, and 4) and five rats from the control group were sacrificed; the bullae (including the middle and inner ear) were isolated from the ears of all rats. Following dissection, the bullae were fixed overnight in a 4% paraformaldehyde solution, and subsequently decalcified using ethylenediaminetetraacetic acid. The specimens were embedded in paraffin, sectioned into 5-μm slices, and stained with hematoxylin and eosin (Sigma-Aldrich).
Statistical analysis
All data was expressed as mean±standard deviation. One-way analysis of variance (ANOVA) was used to determine the statistically significant differences between the control and experimental groups at each time or dose point. Scheffe F-test was used to correct for multiple comparisons when statistically significant differences were identified in the ANOVA. A P-value of <0.05 for the null hypothesis was accepted as indicating a statistically significant difference. Statistical analyses were performed using SPSS ver. 12.0 (SPSS Inc., Chicago, IL, USA).
Go to :
RESULTS
Cell viability assay
The effect of ASD on HMEEC viability after exposure to ASD for 24 hours was initially investigated. Microscopic evaluation showed higher cell debris in the ASD-treated groups (100 and 400 μg/mL) than in the control HMEECs; in addition, there was a sharp decrease in the viable cell population in the group treated with 400 μg/mL of ASD (Fig. 1).
 | Fig. 1.Microscopic images of human middle ear epithelial cell morphology after exposure to Asia sand dust (ASD) for 24 hours. (A) The control group showed 80% of confluence and unified morphology. In contrast, the ASD-treated groups (B, 100 μg/mL; C, 200 μg/mL; and D, 400 μg/mL) demonstrated ASD particles on the cells and many cells were detached from the culture plate in the ASD groups, compared to what was observed in the control group.
|
The CCK-8 cell viability assay demonstrated that ASD caused a dose-dependent decrease in cell viability. Cell viability was considerably decreased in groups exposed to higher concentrations of ASD (>100 μg/mL). Especially, administration of 400 μg/mL of ASD resulted in approximately 60% of viability compared to that of the control group (Fig. 2).
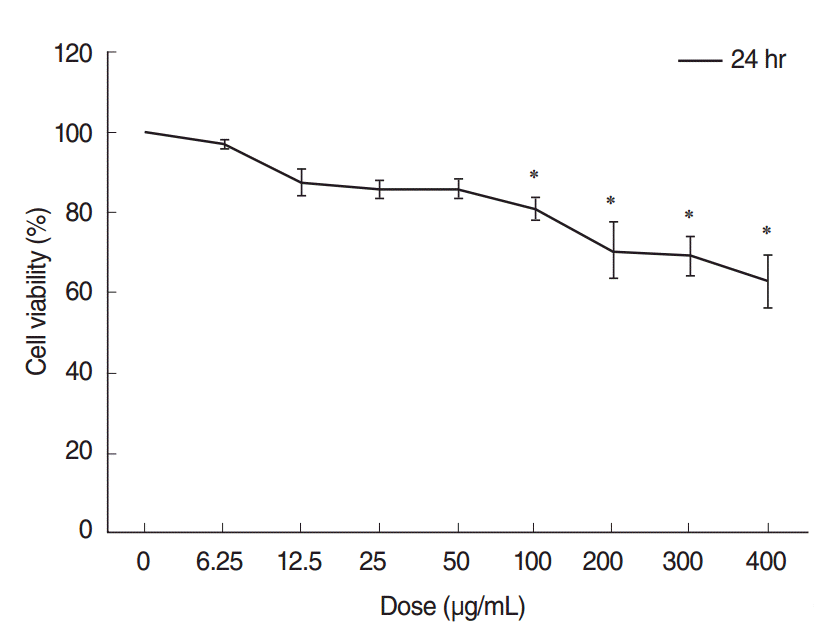 | Fig. 2.Cytotoxicity testing of human middle ear epithelial cells (HMEECs) exposed to Asia sand dust (ASD). Cells were treated with the indicated concentrations of ASD for 24 hours. Cell viability was calculated as the percentage of the viability of the control. HMEEC viability decreased in a dose dependent manner (0–400 μg/mL) when treated with ASD. Especially, administration of 400 μg/mL of ASD resulted in approximately 60% of viability compared to that of the control group. The results presented in the graph are from three independent experiments; the error bars indicate mean±SD. *P<0.05 compared to the control by analysis of variance.
|
Real-time PCR analysis
The expression of TNF-a, COX-2, MUC5AC, and MUC5B genes was analyzed by quantitative RT-PCR, in order to determine whether ASD induces inflammation and mucin production in HMEECs. These genes are usually activated in patients suffering from OM. Exposure to ASD at concentrations greater than 100 μg/mL led to significantly higher mRNA levels of TNF-a and COX-2 (P<0.05) compared to those in the control group (not treated with ASD) (Fig. 3A). ASD exposure also resulted in a prominent increase in the expression of genes coding for the mucin family of proteins, including MUC5AC and MUC5B (P<0.05) (Fig. 3B). In particular, ASD exposure resulted in a significantly higher TNF-a and MUC5B expression compared to that of COX-2 and MUC5AC, respectively (Fig. 3).
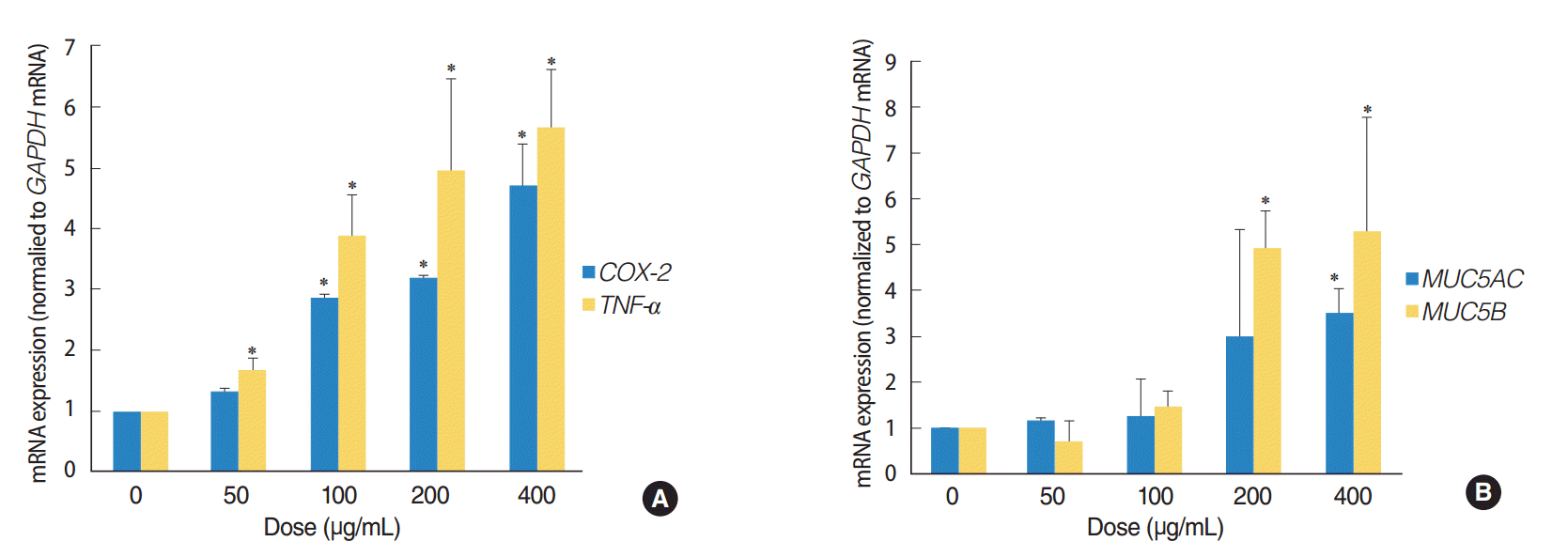 | Fig. 3.Asia sand dust (ASD) stimulates the expression of the inflammation and mucin production genes in human middle ear epithelial cells (HMEECs). HMEECs treated with ASD for 24 hours were subjected to quantitative real-time polymerase chain reaction analysis. HMEECs displayed an increase in inflammatory response and mucin gene expression upon stimulation with ASD in a dose-dependent manner. (A) TNF-a and COX-2 gene expressions were elevated significantly after treatment with ASD at concentrations greater than 100 μg/mL. (B) Treatment with ASD at concentrations greater than 200 μg/mL resulted in a significant increase in MUC5AC and MUC5B gene expressions. The error bars indicate mean±SD of multiple repetitive experiments. *P<0.05 compared to the control, determined by analysis of variance.
|
In addition, we investigated the possible activation of apoptosis and reactive oxygen species (ROS) reactions in HMEECs treated with ASD. Interestingly, the pro-apoptotic gene including TP53 and BAX (P<0.05) were significantly increased, whereas the BCL-2 (P<0.05) mRNA, which is antiapoptotic gene was significantly decreased in HMEECs when stimulated with 400 μg/mL of ASD (Fig. 4). Moreover, the expression of NOX4 and SOD1 genes, which are related with oxidative reaction, were also significantly increased in HMEECs treated with 400 μg/mL of ASD (P<0.05) (Fig. 4). Taken together, these results suggest that ASD induces inflammation and mucin production in HMEECs. Additionally, ASD also induces apoptosis and the production of ROS in HMEECs.
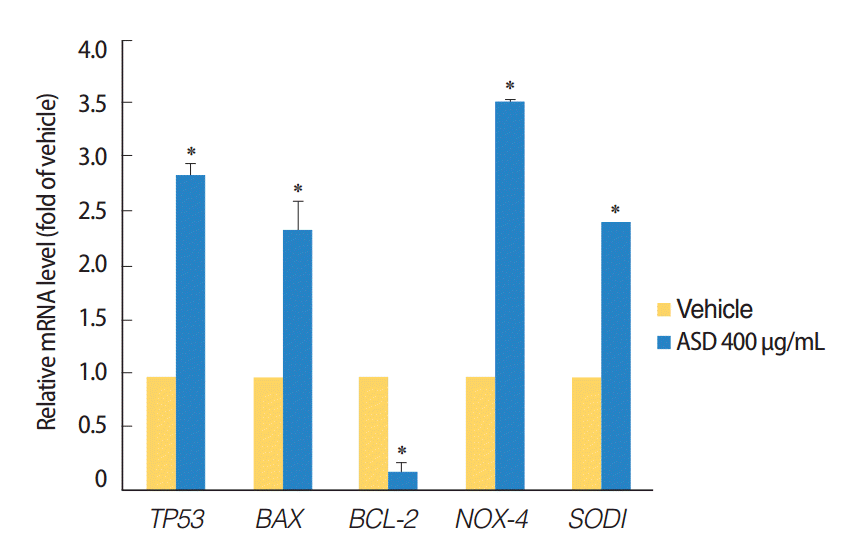 | Fig. 4.Expression levels of the apoptosis and reactive oxygen species production marker genes were investigated. The treatment of Asia sand dust (ASD) (400 μg/mL) for 24 hours resulted in the elevation of TP53 and BAX gene expressions. In contrast, the mRNA levels of the antiapoptotic gene BCL-2 were significantly down-regulated when treated with 400 μg/mL of ASD. ASD also induced an increase of NOX4 and SOD1 gene expressions in human middle ear epithelial cells. The error bars indicate mean±SD of multiple repetitive experiments; *P<0.05 compared to the control by analysis of variance.
|
Western blot analysis
The change in COX-2 protein expression as a result of ASD exposure was also investigated. Western blot analysis demonstrated that ASD treatment significantly enhanced COX-2 expression in HMEECs (Fig. 5). This result suggests that the expression of COX-2 protein is up-regulated in HMEECs exposed to ASD.
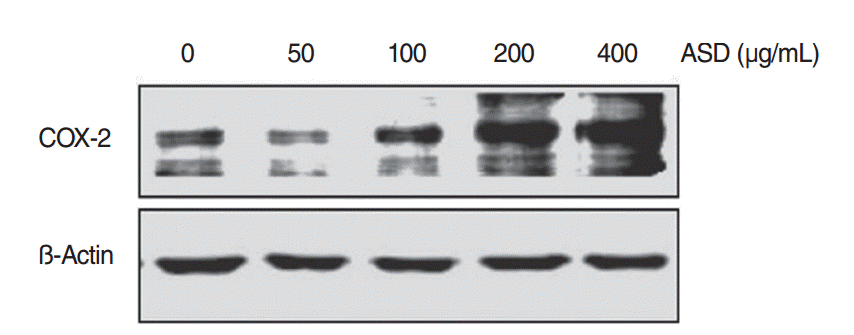 | Fig. 5.The levels of COX-2 protein increased when exposed to Asia sand dust (ASD) for 24 hours. Human middle ear epithelial cells were treated with ASD for 24 hours, at various concentrations (0, 50, 100, 200, or 400 μg/mL) and western blot analysis showed an increase of expression of COX-2 proteins in a dose-dependent manner. Results were obtained from three independent experiments. Beta-actin was used as the loading control.
|
Histopathological examination of the middle ear
The morphology of middle ear epithelium was observed at different time points after ASD treatment by light microscopy. Normal middle ear epithelium was thin and free of inflammatory cells as seen in the control groups (Figs. 6A, 7A). However, when rats exposed to ASD (ASD-injection), the signs of inflammation appeared. At day 1, there was a thickening of the middle ear epithelium and the infiltration of inflammatory cells in the upper region of the epithelium (Figs. 6B, 7B). At day 3, there was a further increase in inflammatory cell infiltration; these cells moved out of the tissue (Figs. 6C, 7C). In addition, we observed cilia in the epithelium of the middle ear (Fig. 7C). However, the mucosal thickness decreased gradually, and inflammatory cell infiltrating the middle ear epithelium reduced in 5 and 14 days after ASD injection (Figs. 6D, E, 7D, E).
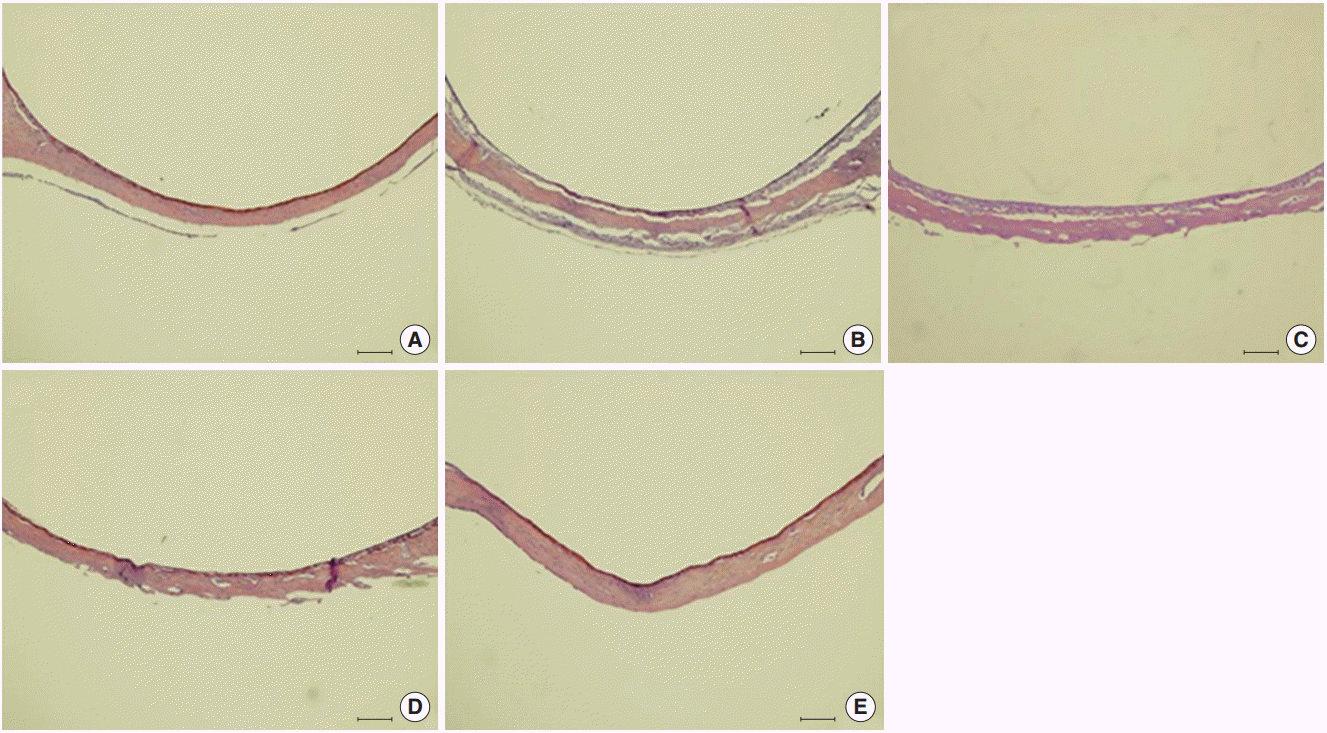 | Fig. 6.Histological changes in the middle ear are observed resulting from Asia sand dust (ASD) injection. Each panel displays representative images of a middle ear cavity at specifi c time points after ASD treatment. (A) shows the control animals (n=10) that received only phosphate buffered saline (vehicle). (B –E) display images of the middle ears taken at 1 (B), 3 (C), 5 (D), and 14 days (E) after ASD injection (each group, n=10). We observed a considerable thickening of the middle ear epithelium on days 1 and 3, which was gradually reduced by day 5. The thickened middle ear mucosa was normalized by day 14 (H&E, ×100, scale bar 100 μm).
|
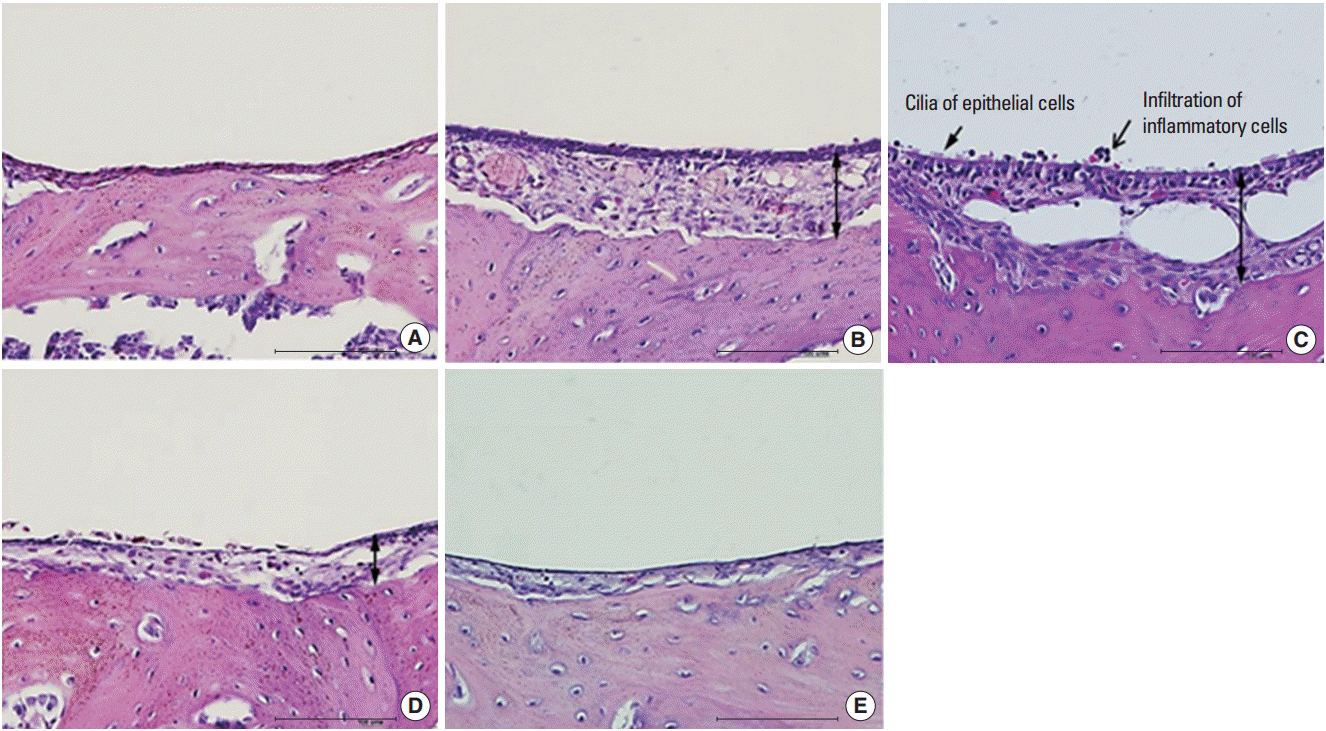 | Fig. 7.Histopathologic finding of otitis media demonstrated in Asia sand dust (ASD) injected rats. The 5 panels display representative images of the middle ear at a specific time point after ASD exposure: (A) control group, (B) day 1, (C) day 3, (D) day 5, and (E) day 14. Images demonstrate the pathological changes occurring as a result of ASD exposure. In particular, 3 days after ASD injection (C), we observed the appearance of cilia in the epithelium of the middle ear (arrow) and a considerable increase in the mucosal thickness (double-headed arrow) in the middle ear cavity. In addition, the middle ear cavity also displayed infiltration of the inflammatory cells (open arrow) (H&E, ×400, scale bar 100 μm)
|
Go to :
DISCUSSION
The major air pollutants include sulfur oxides, nitrogen dioxides, ozone, and inhalant PM. Many previous studies have demonstrated that air pollutions cause adverse effects on human health, such as respiratory inflammation, asthma, heart disease, and lung cancer [6,10]. Interestingly, the biological responses to air pollutions are reported to be different depending on the type of pollutant and degree of exposure [11]. For example, PM can induce oxidative stress in the bronchial epithelial cells, leading to increased susceptibility to infection; diesel exhaust particle, a major air pollutant originating from combustion in motor vehicles, is linked to an acute vascular dysfunction and enhanced thrombus formation.
ASD has become a serious concern in East Asia over the past decade as it can cause diverse harmful effects on human health; this is particularly of concern as ASD is characterized by long-range transport, and can affect an increasing proportion of the population each year. Several epidemiological studies have shown that the annual ASD event to be responsible for numerous negative effects on human health, including cardiovascular and respiratory problems [6,7]. ASD is also known to cause eosinophil infiltration and inflammation in an allergic murine model [12]. However, the correlation between the specific biological signaling network for ASD-induced toxic response and human disease is poorly understood.
Since the cause of OM is multifactorial and includes environmental factors such as smoking and air pollution [1], there are many studies investigating the relationship between ambient air pollution and OM [3,13]. In this study, we have demonstrated that ASD decreased cell viability, and increased the expression of inflammation-related genes (COX-2 and TNF-a) and mucin-producing genes (MUC5AC and MUC5B) in HMEECs. In addition, we observed a significant increase in the mRNA levels of proapoptosis related genes (TP53 and BAX) and ROS producing genes (NOX4 and SOD1) in HMEECs stimulated with ASD. We also observed signs of inflammation in animals exposed to ASD.
The inflammatory response in the middle ear significantly contributes to the development and progression of OM. Many studies have implicated tumor necrosis factor-α (TNF-α) and cyclooxygenase 2 (COX-2) as a key inflammatory cytokines in OM [14,15]. TNF-α is one of the first cytokines that initiates inflammation in the middle ear; it induces the inflammatory response and up-regulates mucin gene expression and mucus secretion in the middle ear [14]. COX-2 is another important proinflammatory mediator that contributes to the increased synthesis of prostaglandins, leading to the prolongation of inflammatory reactions in the middle ear in OM [15]. It is known that TNF-α controls the up-regulation of COX-2 expression and the inflammatory intracellular signaling mechanism [16]. In this study, we showed that ASD induced the classic inflammatory signaling cascade in HMEECs, which is mediated by the TNF-α dependent COX-2 activation pathway.
Mucins, a family of high molecular weight, heavily glycosylated proteins, are produced by mucous glands in the epithelial tissues and perform various functions, from lubrication to cell signaling [17]. Mucin is an important contributor in the development of chronic lung diseases and is overexpressed in asthma, bronchitis, chronic obstructive pulmonary disease, and cystic fibrosis [18]. MUC5AC is the major type of mucin protein found in cases of hyper-secretion of the mucus in the respiratory tract; in addition, MUC5B is known to be related with mucociliary clearance [19], and has been identified in human middle ears affected with chronic otitis media (COM) [20]. In our previous study, we identified that MUC5AC expression is up-regulated in cigarette smoke-induced OM [21]. In current study, we demonstrated that ASD exposure led to an increase in MUC5AC and MUC5B expression. However, the level of expression of the MUC5B gene was higher than that of the MUC5AC mRNA in ASD-exposed HMEECs indicating that mucociliary clearance are more activated upon the exposure to ASD. Also, this implies that the different physical properties and chemical compositions of air pollutants may result in differential mucin gene expression in the middle ear. Moreover, we observed that ASD exposure led to the increased levels of apoptosis- and ROS-related genes, suggesting that apoptosis and ROS formation may be related to increased inflammation and mucin production.
We also explored the effect of ASD on middle ear pathology in vivo using an animal model. It was observed that ASD enhanced the inflammatory response of the middle ear epithelium, as evidenced by the increased mucosal thickness, presence of the cilia on the epithelium, and the influx of inflammatory cells to the specific location.
In this report, we have demonstrated the effects of ASD on the middle ear using HMEECs (in vitro) and an animal model (in vivo). However, this study is limited in that only the biological effects of ASD on HMEECs, such as inflammation, mucin production, apoptosis, and ROS production, were determined. Therefore, further studies should focus on investigating the gene expression profiles in order to determine the specific regulatory mechanisms activated by ASD exposure. This may help understand the development of ASD-induced OM.
In conclusion, ASD caused a decrease in cell viability and increase the inflammation, mucin production, apoptosis, and oxidative stress-related genes (mRNA) in HMEECs. Additional studies conducted in animal model clearly demonstrated that ASD exposure caused an inflammatory response in the middle ear of rats. Overall, these findings demonstrated that exposure to ASD can cause damage to the middle ear epithelium, and induce an inflammatory responses. In addition, our data indicates that ASD may be a risk factor for OM, which provides an important link between environmental air pollution and middle ear diseases.
Go to :
HIGHLIGHTS
▪ We evaluated the effect of Asia sand dust (ASD) in human middle ear epithelial cells (HMEECs).
▪ ASD exposure decreased the HMEEC viability dose-dependently.
▪ ASD exposure significantly increased inflammation-related genes (COX-2, TNF-a).
▪ ASD exposure significantly increased mucin-pro ducing genes (MUC5AC, MUC5B).
▪ Environmental ASD exposure can result in the development of otitis media.
Go to :
 XML Download
XML Download