

 PDF
PDF Citation
Citation Print
Print



서 론
발암 과정에 있어 정상 세포가 여러 가지 생물학적인 능력들을 갖게 되고, 이러한 암세포의 특성들(hallmarks of cancer)은 암의 종류를 막론하고 공통적으로 보이는 현상으로 간주된다[1]. 특히, 세포 에너지 활용의 변화 혹은 에너지대사의 재편성은 최근에 추가된 암의 중요 특성으로서, 암이 그 종류와 원인이 다양하게 존재함에도 불구하고, 이러한 성향은 모든 암에서 공유된다[2]. 암세포 대사의 특징적인 변화로 대표적으로 거론되는 것이 와버그 효과(Warburg effect)이다. 이것은 암세포가 산소가 충분히 존재하는 조건임에도 불구하고 에너지 대사적으로 효율이 높은 산화적 인산화(oxidative phosphorylation, OXPHOS) 과정보다 해당 과정(glycolysis)을 선호하는 현상을 일컫는 것으로서 호기성 해당 작용(aerobic glycolysis)으로도 불린다[3]. OXPHOS 과정을 통해 많은 양의 adenosine triphosphate(ATP)(포도당 한 분자당 36개의 ATP)를 생산하는 대신 상대적으로 에너지 효율이 떨어지는 해당 과정(포도당 한 분자당 2개의 ATP)을 채택함으로써 암세포는 보다 빠르게 ATP를 생산하고, 활성 산소종(reactive oxygen species, ROS)의 발생을 줄이고, 저산소의 종양미세환경(tumor microenvironment)에서 당대사의 안정성을 갖추고, 중요하게는 당의 중간대사체가 여러 생합성 경로로 유입되는 것을 늘려, 궁극적으로 암세포 성장에 필요한 뉴클레오티드(nucleotide), 아미노산, 지방 등의 바이오매스(biomass) 합성을 통한 동화작용(anabolism)에 기여하게 되는 생물학적인 장점을 가지게 된다.
약 100여 년 전 Warburg 등[4]에 의해 본 이론이 처음 제기될 당시에는, 암세포 내에서는 미토콘드리아의 기능 부전으로 인해 OXPHOS 과정이 손상되거나 억제되고 있다고 생각을 하였다. 그러나 이후에 암세포 내에서도 정상 세포와 마찬가지로 OXPHOS 과정이 일어날 수 있다고 알게 되었으며[5-8], 암세포가 놓인 다양한 환경에 따라 해당 작용과 OXPHOS 작용 간에 대사 전환(metabolic switch)을 통해 능동적으로 대처할 수 있음을 알게 되었다. 암세포 내에서는 당 흡수 증가를 통해 당대사가 활성화되어 있지만, 당 이외의 다른 영양분 흡수를 통해 미토콘드리아 내 tricarboxylic acid(TCA) 회로가 활성화되기도 한다.
과거에는 암생물학에 있어 암대사(cancer metabolism)가 주류 학문으로서 큰 관심을 받지 못하다가, 최근에 들어서 암대사가 종양신호전달(oncogenic signal transduction)이나 종양면역(tumor immunity)과 마찬가지로 암생물학의 필수 요소로 그 관심이 증가하고 있다. 특히나 암대사 중에서도 지방산 대사와 관련하여 그 역할에 대해 재조명되고 있어, 이 분야에 대한 보다 깊은 이해를 통해 두경부암의 치료에 대한 새로운 접근을 해 볼 수 있을 것이다. 본 종설에서는 특히 지방산의 합성, 산화, 그리고 저장에 초점을 맞추어 암세포 내에서 보이는 대사에 대해 살펴보고, 이를 통해 암의 증식과 진행을 억제할 수 있는 전략들을 모색해보고자 한다.
Go to : 
본 론
암세포의 특이적 당대사(Characteristic glucose metabolism in cancer cells)
암세포는 증식과 성장을 위해서 당대사(glucose metabolism)를 활용하게 되는데, 이는 여러 암 돌연변이에 따른 신호 전달의 강화를 통해 일어나게 된다. 이러한 현상을 임상적으로 적용한 예가 암세포가 포도당의 유사체인 18F-fluorodeoxyglucose 를 흡수함으로써, 암의 진단 및 치료 전후 반응성 평가 등에 활용할 수 있는 검사인 양전자방출단층촬영술(positron emission tomography, PET)이다. 여하튼 암세포의 여러 구성 요소(building block)의 합성이 필요한 상황에서, 세포 내로 흡수된 포도당이 오로지 미토콘드리아 내의 OXPHOS 과정을 통해 분해되어 많은 양의 ATP를 생산하는 것 만으로는 도움이 되지 않고, 포도당의 고분자 전구물질로의 전환이 필수적인데, 가령 지방산 합성을 위한 acetyl-CoA, 비필수 아미노산 합성을 위한 해당 중간체(glycolytic intermediates), 그리고 뉴클레오티드의 합성을 위한 라이보오스(ribose)의 생성이 그것이다(Fig. 1).
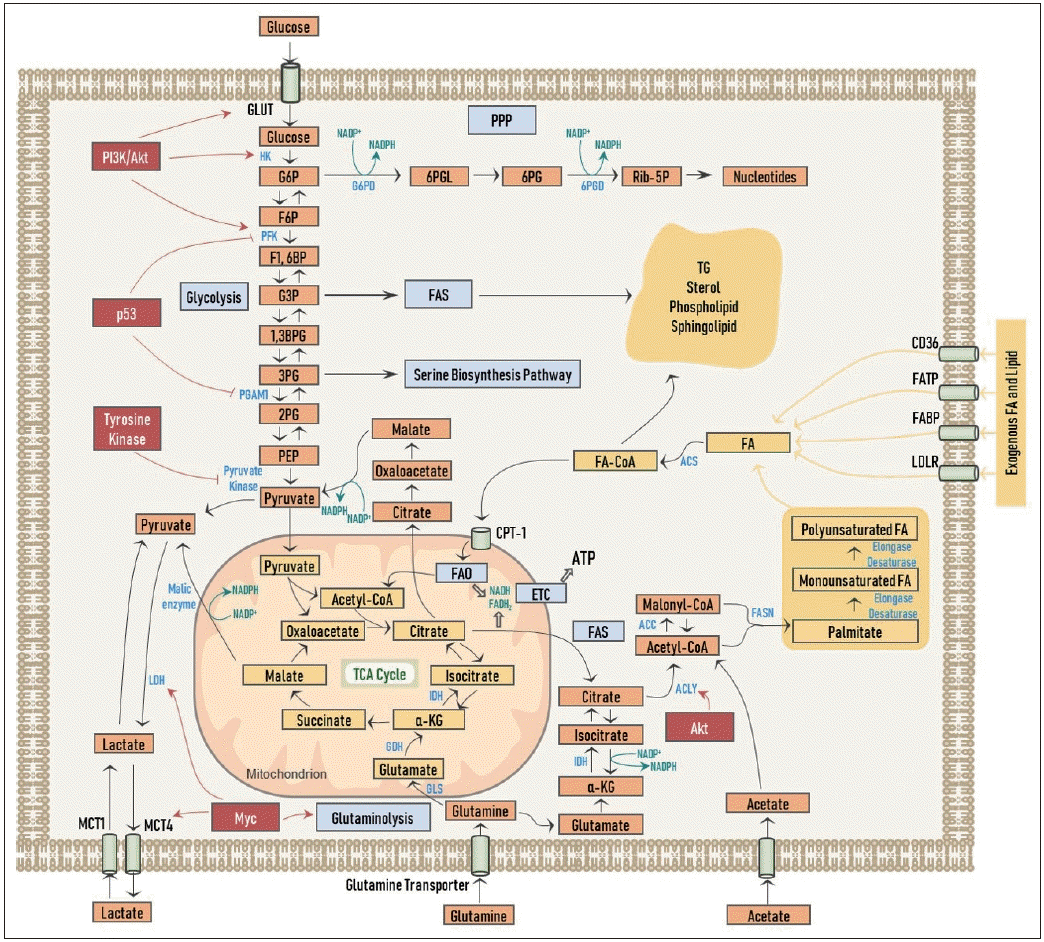 | Fig. 1.Overview of cancer cell metabolism. Regulation of metabolism by oncogenic signaling pathways are also shown. See text for detailed description of depicted pathways. GLUT: glucose transporter, MCT1: monocarboxylate transporter1, MCT4: monocarboxylate transporter4, FATP: fatty acid transport protein, FABP: fatty acid binding protein, LDLR: low-density lipoprotein receptor, CPT-1: carnitine palmitoyl transferase-1, G6P: glucose-6-phosphate, F6P: fructose-6-phosphate, F1,6BP: fructose-1,6-biphosphate, G3P: glyceraldehyde-3-phosphate, 1,3BPG: 1,3-biphosphoglycerate, 3PG: 3-phosphoglycerate, 2PG: 2-phosphoglycerate, PEP: phosphoenolpyruvate, 6PGL: 6-phosphogluconolactone, 6PG: 6-phosphogluconate, Rib-5P: ribose-5-phosphate, α-KG: α-ketoglutarate, HK: hexokinase, PFK: phosphofructokinase, PGAM1: phosphoglycerate mutase-1, LDH: lactate dehydrogenase, G6PD: glucose-6-phosphate dehydrogenase, 6PGD: 6-phosphogluconate dehydrogenase, IDH: isocitrate dehydrogenase, GDH: glutamate dehydrogenase, GLS: glutaminase, ACLY: ATP-citrate lyase, ACC: acetyl-CoA carboxylase, FASN: fatty acid synthase, ACS: acyl-CoA synthetase, PPP: pentose phosphate shunt, FAS: fatty acid synthesis, FAO: fatty acid oxidation, ETC: electron transport chain, TCA: tricarboxylic acid, TG: triacylglycerol, FA: fatty acid, NADP: nicotinamide adenine dinucleotide phosphate (oxidized form), NADPH: nicotinamide adenine dinucleotide phosphate (reduced form), ATP: adenosine triphosphate, FA-CoA: fatty acid-CoA.
|
각종 성장 인자 및 PI3K/Akt 등의 종양 신호 전달에 의해 포도당 수송체가 활성화되어 포도당 세포 내로의 흡수가 증가되고, 헥소키나아제(hexokinase), 포스포프룩토키나아제(phosphofructokinase, PFK)를 비롯한 해당 작용의 주요 효소들의 기능 조절을 통해 해당 작용은 강화된다[9,10]. 더 나아가, 타이로신인산화효소(tyrosine kinase) 성질을 가지는 많은 수의 암유전자들은 피루브산염 키나아제(pyruvate kinase)의 활성을 특이적으로 억제하여 이러한 일련의 현상들이 당중간대사체인 포도당-6-인산(glucose-6-phosphate, G6P)이 펜토오스 인산 경로(pentose phosphate shunt, PPP)로의 유입을 늘려 뉴클레오티드 합성을 용이하게 한다(Fig. 1). 이 과정에서 포도당-6-인산탈수소효소(G6P dehydrogenase, G6PD)와 6-포스포글루콘산탈수소효소(6-phosphogluconate dehydrogenase, 6PGD) 두 효소에 의해 nicotinamide adenine dinucleotide phosphate(NADPH) 또한 생성되는데, 이를 통해서 합성에 필요한 전자(electrons)를 전달받게 되고, 또한 산화 환원 조절(redox control)을 할 수 있게 된다. 뿐만 아니라 해당 중간체 중의 하나인 3-인글리세르산(3-phosphoglycerate)의 생성 역시 증가되어 세린 생합성 경로(serine biosynthesis pathway)를 통해 비필수 아미노산 생성을 늘리게 된다. 암억제 인자로 잘 알려진 p53은 PFK와 인산글리세르산변위효소-1 (phosphoglycerate mutase-1)을 억제하여 해당 중간체가 각각 PPP와 세린 생합성 경로로의 유입을 증가시킨다(Fig. 1). 또한 지방산 합성을 위해 필요한 탄소는 포도당의 acetyl-CoA로의 전환을 통해서 얻을 수 있게 된다.
해당 작용을 통해 생성된 피루브산염은 젖산탈수소효소(lactate dehydrogenase, LDH)를 통해 젖산(lactate)으로 변환되고, 이로써 과다한 탄소는 배출되고 또한 NADPH의 생성을 돕는다. 젖산은 모노카복실레이트 수송체(monocarboxylate transporter, MCT)의 하나인 MCT4를 통해 암세포 밖으로 배출되지만, 일부 암세포에 따라서는 거꾸로 MCT1을 통해 다시 암세포 내로 흡수하여 미토콘드리아 내 OXPHOS 과정에 이용된다는 연구도 보고된 바 있다[11]. 암세포에서는 Myc를 통해 LDH와 MCT의 발현이 증가되어 있다[12]. Myc는 또한 글루타민(glutamine)의 세포 내 흡수와 분해(glutaminolysis)를 활성화시켜 글루타민산(glutamate)이 말린산염(malate)과 피루브산염(pyruvate)을 거치면서 NADPH의 생성을 촉진시킨다(Fig. 1).
대사의 재편성(Reprogramming of metabolism)
성장하는 암세포에서 해당 과정을 증식성 대사(proliferative metabolism)로 활용하는 것에 반해, 전이성 침습성 암세포에서는 다른 방향으로 대사가 재편성된다(Fig. 2). 이동 중인 전이암세포에서는 포도당의 흡수가 감소되고, 이로 인해 PPP로부터의 NADPH 생산이 감소되어 세포 내 산화 환원 능력이 떨어져 ROS 발생이 증가하게 된다. 게다가 ATP 생산 또한 감소하게 되어 이러한 일련의 에너지, 산화적 스트레스(energetic and oxidative stress)로 인해 암세포는 세포사의 위기에 놓이게 된다. 그러나 그에 따라 암세포 내에서는 ATP의 감지자(sensor)인 adenosine monophosphate(AMP)-활성 단백질 인산화효소(AMP-activated protein kinase, AMPK)가 활성화되고 acetyl-CoA 카복실화효소(acetyl-CoA carboxylase, ACC)가 억제되어 지방산 합성(fatty acid synthesis, FAS)은 억제되고 지방산 산화(fatty acid oxidation, FAO)는 활성화된다. FAS가 감소함으로써 NADPH 소모량은 줄어들고, FAO가 증가함으로써 말린산염에서 피루브산염으로의 전환과 아이소구연산염(isocitrate)에서 알파-케토글루타르산(α-ketoglutarate, α-KG)로의 전환이 각각 활성화되고, 이 과정에서 NADPH 생산이 증가하여 부족한 양을 보충할 수 있게 된다. 또한 FAO를 통해 미토콘드리아 내 OXPHOS를 통해 많은 양의 ATP를 생산하게 된다. 이처럼 침습성 대사(invasive metabolism)에서는 지방산의 대사가 중요한 역할을 한다.
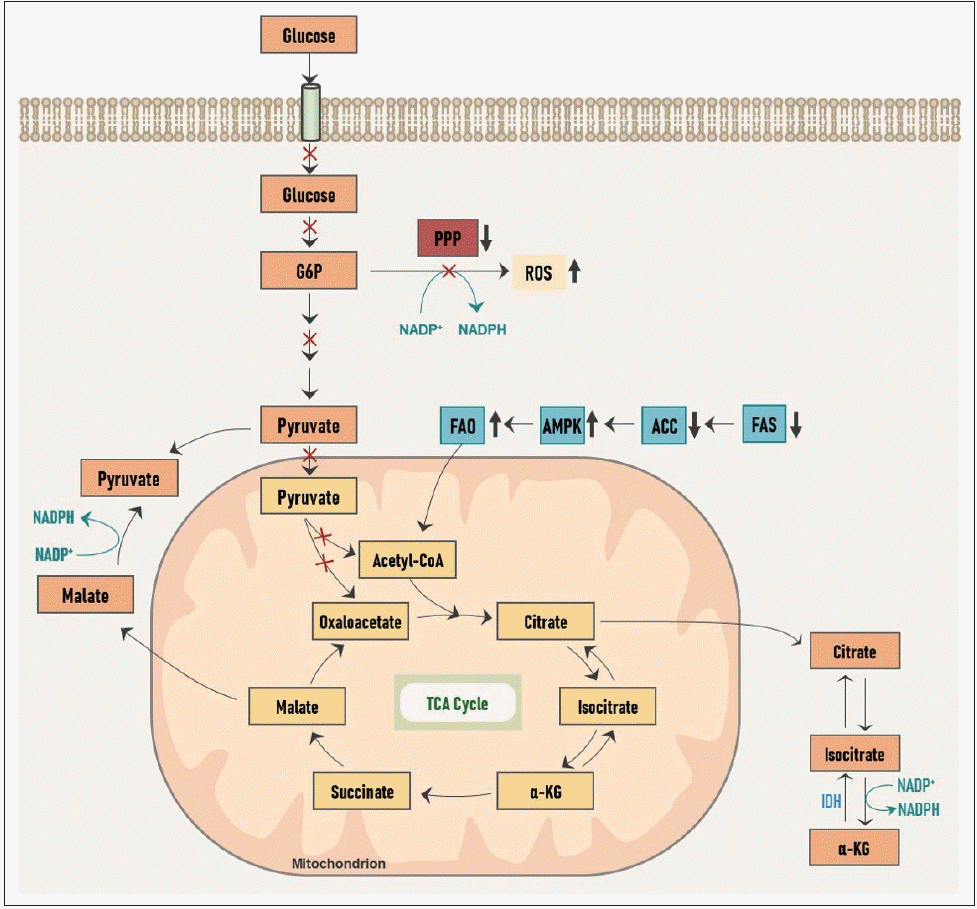 | Fig. 2.Reprogramming of metabolism in metastatic, invasive cancer cells. Cancer cells during migration and invasion undergo alterations in metabolism in order to survive the energetic and oxidative stress. Lipid metabolism has a central role during invasive metabolism of cancer. G6P: glucose-6-phosphate, α-KG: α-ketoglutarate, PPP: pentose phosphate shunt, FAO: fatty acid oxidation, AMPK: AMP-activated protein kinase, AMP: adenosine monophosphate, ACC: acetyl-CoA carboxylase, FAS: fatty acid synthesis, ROS: reactive oxygen species, TCA: tricarboxylic acid, IDH: isocitrate dehydrogenase, NADP: nicotinamide adenine dinucleotide phosphate (oxidized form), NADPH: nicotinamide adenine dinucleotide phosphate (reduced form).
|
암세포에서의 지방대사(Lipid metabolism in cancer cells)
지질은 다양한 군으로 이루어진 소수성의 생분자물질로서 인지질(phospholipids), 스핑고지질(sphingolipids), 트리아실글리세롤(triacylglycerols, TG), 스테롤(sterols) 등을 포함하며 지방산이 기본 합성단위가 된다. 지방산은 다양한 탄소의 수를 가지는 탄화수소 사슬이 그 말단에 카복시기(carboxyl group)가 붙어 있는 구조로 이루어져 있으며 다양한 포화도를 보인다. 세포 내에서 지방산은 세포막의 구성 성분이 되기도 하고, 에너지 대사 및 저장, 그리고 신호전달물질로써도 활용이 된다. 암세포에서의 지방산 대사 중 대부분 연구의 초점은 주로 FAS에 맞추어졌으나, 최근 들어 암에서의 FAO의 역할에 대해서도 관심이 증가하고 있다.
지방산 합성(FAS)
암세포가 지방산을 얻는 방법은 세포 외부로부터 흡수를 하거나, 세포 내에서 직접 합성을 하는 2가지 경로로써 가능하다(Fig. 1). 정상 세포에서는 주로 지방산의 외인성 흡수를 통해 지방 필요량을 보충하는 데 반해서 암세포에서는 상대적으로 지방산의 신생합성(de novo FAS)이 활성화되어 있는 것으로 알려져 있다. 그러나 최근에는 암세포에서도 주변 환경의 지방산을 필요에 따라 활발히 흡수하고, 암의 진행에 있어 이러한 외인성 지방산 흡수와 지방산의 신생합성 모두가 중요하다는 연구 결과들이 늘고 있다[13,14]. 특히, 암세포는 필요에 따라 주위 종양미세환경의 지방세포(adipocyte)로부터 유리된 지방산을 흡수하여 지방대사에 활용할 수 있다[15-18]. 지방산을 세포 내로 흡수하는 역할을 담당하는 대표적인 수송체로는 CD36, 지방산수송단백질(fatty acid transport proteins, FATPs), 지방산결합단백질(fatty acid binding proteins, FABPs), low-density lipoprotein receptor 등이 있다.
FAS의 첫 단계는 구연산염(citrate)으로서, 이는 주로 앞서 살펴본 해당 과정의 산물인 피루브산염이 TCA 회로를 경유하여 생성된다. 그러나 저산소 환경이나 미토콘드리아의 기능이 억제되어 있는 경우에 TCA 회로가 돌아갈 수 없게 되어, 암세포는 대체 영양소로서 글루타민을 세포 내로 흡수하여 α-KG를 생성하고, 아이소구연산염 탈수소효소(isocitrate dehydrogenase, IDH)에 의한 환원성 카복시화 반응(reductive carboxylation)를 통해 구연산염을 생성하게 된다[19]. 구연산염은 이어서 ATP-구연산염 분해효소(ATP-citrate lyase, ACLY)에 의해 acetyl-CoA로 전환되는데, 암세포에서는 Akt를 통해 ACLY의 활성화를 촉진시켜 acetyl-CoA의 합성을 증가시킨다[20]. 암세포는 때로는 MCT를 통해 외부에서 아세트산염(acetate)을 흡수하여 acetyl-CoA를 직접 생성하기도 한다. 해당 과정이 상대적으로 저조하고 지방산 대사가 활성화되어 있는 암에서는 이러한 현상을 이용하여 영상학적인 진단 검사가 가능한데, C11-acetate PET 검사가 그 예이다[21]. Acetyl-CoA는 이어서 속도제한효소(rate-limiting enzyme)인 ACC에 의해 malonyl-CoA로 전환된다. Malonyl-CoA로부터 지방산 합성효소(fatty acid synthase, FASN)에 의해 비로소 포화지방산인 팔미테이트(palmitate)가 생성이 된다. 팔미테이트는 이후에 여러 신장효소(elongase)와 불포화효소(desaturase)의 작용에 의해 다양한 탄소의 수와 포화도를 가지는 지방산들을 생성하게 된다. 이와 같이 생성된 지방산들은 아실-CoA 합성효소(acyl-CoA synthetase)에 의해 fatty acid-CoA로 가공이 되어 세포막의 구성 성분으로 중요한 인지질이나 스핑고지질로 전환되거나, TG와 스테롤로 바뀌어 지방 방울(lipid droplet, LD)의 형태로 저장이 된다. 에너지 저장소인 LD는 추후에 암세포가 필요시 FAO의 연료로서 공급하게 된다.
ACLY, ACC, FASN 등 FAS에 관여하는 주요 효소들은 스테롤 조절 요소 결합 단백질-1(sterol regulatory element binding protein-1, SREBP-1)이라는 전사조절인자에 의해 발현되고, SREBP는 PI3K/Akt/mTORC 종양 신호에 의해 활성화된다.
세포 내에서는 포화지방산 대비 불포화지방산의 비율이 일정하게 유지되는 것이 중요한데, 포화지방산이 과도하게 쌓이게 되면 이로 인해 미토콘드리아의 기능 이상, ROS 증가, 소포체(endoplasmic reticulum, ER) 스트레스 등이 발생하여 세포에 치명적인 손상을 가져다줄 수 있게 된다. 따라서 지방산의 탈포화(desaturation)를 유도하는 스테아로일-CoA 불포화효소(stearoyl-CoA desaturase, SCD)의 역할이 중요한데, 이 효소는 특이적으로 산소가 충분한 환경에서 작용이 원활하게 이루어진다. 암세포에서는 SCD의 발현이 증가되어 있는 것으로 알려져 있으며[22,23], 저산소 환경에 놓인 암세포에서는 SCD의 활성이 억제되어 불포화지방산의 생성이 감소하기에, 이러한 상황에 대비하여 암세포는 LD 내의 불포화지방산의 유리를 증가시키거나[24] 저산소증유발인자-1α(hypoxiainducible factor-1α) 발현 증가를 통해 외부 환경으로부터 불포화지방산의 흡수를 늘려서 지방독성(lipotoxicity)으로부터 세포를 보호하게 된다[25].
포화지방산과 불포화지방산의 세포 조성은 세포막의 유동성(fluidity)에도 깊은 영향을 미친다. 즉, 포화지방산의 비율이 높을수록 세포막은 견고해지고 유동성은 감소하게 되고, 이러한 경우 암세포는 약물 흡수 감소로 인해 항암제로부터 저항성을 가질 수 있게 된다[26]. 반면에, 불포화지방산의 비율이 높은 암세포에서는 세포막의 구성이 느슨해져 유동성은 증가하여 이로 인해 암세포의 이동과 전이를 용이하게 한다[27]. 이처럼, 지방산의 조성에 따른 세포막의 유동성을 조절하여 암세포는 각 필요 상황에 맞도록 이점을 취할 수 있게 된다.
지방산 산화(FAO)
앞서 언급한 바와 같이, 전이성 암세포에서는 포도당의 흡수가 줄어들고 암세포의 이동과 침습의 과정에서 많은 양의 ATP를 필요로 하게 되는데, 이러한 상황에서 가장 효율적으로 에너지를 생산할 수 있는 방법이 FAO이다. 탄수화물과 비교해서 지방산은 2배나 많은 양의 ATP를 제공할 수 있다. 두경부암세포 중에서도 전이를 유발하는 일부 세포들에서 지방산의 수용체인 CD36의 발현과 함께 지방대사 관련 유전자의 발현이 높은 것을 확인한 연구가 있으며[13], 이 특정 암세포군에 팔미테이트 혹은 고지방식이(high fat diet) 처리 시에 원발 부위 암의 변화는 미미하고, 특이하게 암세포의 전이만을 촉진시켰고, 반대로 CD36을 억제 시에 원발 부위의 암에 대한 영향없이 암의 전이만 선택적으로 억제할 수 있음을 확인한 바 있다. 뿐만 아니라, 원발 부위의 암과 비교하여 림프절로 전이가 이루어진 암세포를 분석한 결과, FAO와 관련된 유전자의 발현이 증가되어 있으며, 이는 특이하게 활성화된 yes연관 단백질(yes-associated protein, YAP)이라는 전사인자에 의해 유도됨을 확인한 연구 결과도 있다[28]. 이 연구에서도 FAO를 억제하는 약물을 투여하거나 YAP 발현을 억제했을 때 림프절 전이를 선택적으로 억제할 수 있었다. 이를 토대로 볼 때 FAO를 억제하는 것이 암의 침습과 전이를 억제할 수 있는 효과적인 전략임을 짐작해 볼 수 있을 것이다.
지방산은 이처럼 CD36을 통해 외부에서 세포 내로 흡수가 되거나 LD 내에 저장되어 있던 TG로부터 유리되어, 카르니틴팔미토일 전이효소(carnitine palmitoyl transferase-1)에 의해 미토콘드리아 내로 이동하여 FAO가 이루어진다. FAO 혹은 베타 산화과정(β-oxidation)의 결과로 acetyl-CoA, nicotinamide adenine dinucleotide(NADH), flavin adenine dinucleotide(FADH2)가 생성되고, acetyl-CoA는 TCA 회로로 유입되어 NADH가 생성되고, NADH와 FADH2는 전자전달계(electron transport chain, ETC)로 유입되어 ATP가 발생하게 된다(Fig. 1). 미토콘드리아 내 acetyl-CoA가 TCA 회로를 통해 NADH를 생성하지만, 함께 구연산염 또한 생성되어 세포질로 유출되고, 이어서 다음의 두 가지 경로로 연결되어 그 과정에서 NADPH가 생성된다: 1) 각각 연속적으로 옥살로아세트산염(oxaloacetate), 말린산염, 피루브산염으로 전환되는 경로, 2) IDH에 의해 아이소구연산염에서 α-KG로 변환되는 경로. 이처럼 생성된 NADPH는 전술한 바와 같이 전이성 암세포에서 ROS 발생 증가로 인한 산화적 스트레스를 감소시키는 항산화제로서의 역할을 하지만, 한편으로는 동화 작용에 관여하는 여러 효소들에 대한 조효소로서 작용하여 증식성 암세포의 합성 과정에도 이용될 수 있다. 요약하자면, 암세포에서는 필요시 FAO의 과정을 통해 ATP와 NADPH를 공급받을 수 있는 것이다[29]. 또한, FAO를 통해 생성된 acetylCoA는 구연산염으로의 변환 과정을 거쳐 세포질 내 acetylCoA로 다시 변환되어 FAS로 이어질 수도 있다는 점을 미루어 보아, FAO와 FAS는 필요에 따라 상호 보완적인 관계로 세포에 유용하게 작용할 수 있다.
지방 대사의 치료 표적(Therapeutic targets in lipid metabolism)
지금까지 살펴봤듯이 암세포 내에서는 특이적인 지방 대사를 통해 외부 환경에 따라 대사적 가소성(metabolic plasticity)을 보이고 있으며, 따라서 암의 이러한 생존 전략을 역이용하여 암치료의 표적으로 모색해볼 수 있을 것이다(Table 1).
Table 1.
Drugs targeting cancer cell specific lipid metabolism
| Target | Drug | Comments | References |
|---|---|---|---|
| FASN | Orlistat | FDA approved | [31,35] |
| Cerulenin | [32,35] | ||
| C75 | [32,35] | ||
| IPI-9119 | [33] | ||
| EGCG | Green tea leaf extract | [34,35] | |
| TVB-3166 | [39] | ||
| TVB-2640 | [40] | ||
| Triclosan | [64] | ||
| ACLY | LY294002 | PI3K inhibitor | [41] |
| SB-204990 | [42] | ||
| ACC | Soraphen A | [43] | |
| TOFA | Also inhibits SCD activity | [44,45,54] | |
| Glutaminase | BPTES | [46] | |
| SREBP-1 | fatostatin | [47,48] | |
| betulin | [49] | ||
| SCD | Betulinic acid | [55] | |
| SSI-4 | [56] | ||
| MF-438 | [57] | ||
| BZ36 | [65] | ||
| CPT-1 | Etomoxir | [28,59] | |
| Perhexiline | [60] | ||
| Oxfenicine | [66] | ||
| 3-KAT | trimetazidine | [61] | |
| ranolazine | FDA approved | [62] | |
| ETC | Metformin | Also activates AMPK | [63] |
| Phenformin | [67] |
FASN: fatty acid synthase, ACLY: ATP-citrate lyase, ATP: adenosine triphosphate, ACC: acetyl-CoA carboxylase, SREBP-1: sterol regulatory element binding protein-1, SCD: stearoyl-CoA desaturase, CPT-1: carnitine palmitoyl transferase-1, 3-KAT: 3-ketoacylthiolase, ETC: electron transport chain, FDA: Food and Drug Administration, PI3K: phosphoinositide 3-kinase, AMPK: AMP-activated protein kinase, AMP: adenosine monophosphate
![]()
지방산 합성의 표적화(Targeting FAS)
외인성 지방산의 세포 내 흡수를 담당하는 막단백질을 치료 표적으로 하는 전략을 통해 암세포의 지방산 공급을 막을 수 있다. 항CD36 항체를 통해 항암 효과가 있음을 확인하였으며[13,14], FATP 및 FABP에 대한 억제 약물을 통해서도 암의 전이를 효과적으로 줄일 수 있음을 확인한 바 있다[15,30].
이와 대별하여 암세포 내의 지방산 신생 합성을 표적으로 하는 치료 전략에 대한 연구들도 활발히 진행 중이다. 정상 세포에 비해 암세포 내에서 지방산의 신생 합성이 상대적으로 활성화되어 있기에 매력적인 항암전략으로서 주목을 많이 받고 있다. 지방산 신생 합성 과정에서 작용하는 효소 중에서 FASN을 억제하는 여러 약물이 개발되어 많은 연구가 있었는데, orlistat, cerulenin, C75, IPI-9119, EGCG가 그 예이다[31-35]. 그러나 동물 실험을 통해 이러한 약을 사용했을 때 급격한 체중 소실이나 신경줄기세포의 기능 이상으로 일부 심각한 부작용이 확인되어 임상 적용에는 아직 세심한 주의가 필요한 상황이다[32,36]. 뿐만 아니라, FAS 억제를 통해 상대적으로 FAO 활성화를 통한 암 전이가 증가되었다는 결과도 있어[37,38], 광범위한 암의 적용보다는 암의 각 진행 단계에 맞는 치료 전략을 시사한다. 그러나 최근에는 FAS 억제를 하지만 간접적인 FAO 활성화 효과를 보이지 않는 TVB-3166이나 TVB2640과 같은 차세대 FASN 억제제가 개발되어, 지방 조직 소실 및 심각한 체중 감소 등의 부작용 없이 보다 특이적인 FAS 억제 효과를 보여 탁월한 항암효과의 가능성을 보인 결과도 있다[39,40]. FASN 이 외에도 ACLY와 ACC를 표적으로 하는 치료 전략에 대해서도 많은 연구가 되어, 임상 적용의 잠재적 가능성을 엿볼 수 있다[41-44]. 특히나 epidermal growth factor receptor 억제제인 cetuximab에 대한 저항성을 보이는 두경부암세포에서 cetuximab과 함께 ACC 억제제인 TOFA를 병용처리하였을 때에 효과가 있었다는 보고도 있다[45]. 뿐만 아니라 암세포는 상황에 따라 대체영양소로서 글루타민을 흡수하여 글루타민대사 과정을 통해 TCA 회로로 유입되거나 구연산염으로 전환되어 FAS로 이어지는데, 글루타민 분해 효소(glutaminase)를 억제하는 약물인 BPTES 처리를 통해 두경부암세포의 성장을 저해할 수 있었다는 결과도 보고된 바 있다[46].
ACLY, ACC, FASN, SCD 등 FA 합성 과정에 관련된 효소들의 발현은 전사조절인자인 SREBP-1에 의해 조절된다. 그러나 SREBP-1는 전사인자이기에 이를 직접 억제하는 약물 개발은 어렵고 SREBP-1과 관련된 단계에서 유용한 치료 전략을 생각해볼 수 있다. SREBP-1이 활성화되기 위해서는 SREBP-1이 SREBP-분열 활성화 단백질(SREBP-cleavage activating protein, SCAP)과 결합하여 ER에서 골지체(golgi apparatus)로 이동을 하게 된다. SREBP-1과 SCAP의 결합을 방해하는 약물이 개발되어 항암효과가 있음을 확인한 바 있는데 fatostatin, betulin 등이 그 예이다[47-49]. 그 밖에도 SREBP-1의 상위 조절인자인 간 X 수용체(liver X receptor)를 억제하는 전략에 대한 연구도 보고된 바 있다[50,51].
요컨대 암세포는 지방산 신생 합성뿐만 아니라 외인성 지방산 흡수를 통해서도 세포가 필요한 지방산 공급을 받을 수 있기에, 이 두 과정을 모두 막는 것이 더 효과적인 치료 전략일 수 있다. 한편, 전술한 바와 같이 지방산 합성을 통해 생성된 팔미테이트는 여러 신장효소와 불포화효소의 작용을 통해 다양한 단일불포화지방산(monounsaturated fatty acid)과 다불포화지방산(polyunsaturated fatty acid)을 생성하여 세포가 필요한 다양한 용도로 활용되는데, 암세포의 신장효소와 불포화효소를 각각 억제하여 암의 증식과 성장을 억제할 수 있다는 결과가 보고된 바 있다[52-54].특히 대표적인 불포화 효소인 SCD에 대한 억제제가 많이 개발되고 연구되었는데, betulinic acid, SSI-4, MF-438 등이 있다[55-57]. 그러나 일부 암세포에서는 SCD 억제제를 처리하였을 때 외인성 지방산 흡수나 다른 불포화효소의 작용을 통해 불포화지방산의 공급을 유지하여 SCD 억제제에 대한 저항성을 보인 경우도 있다[25,58].
지방산 산화의 표적화(Targeting FAO)
FAO의 가장 첫 단계이자 속도 조절 단계(rate-limiting step)인 CPT-1의 작용을 억제하는 약제가 개발되어 암의 성장 및 진행을 제어할 수 있다는 결과들이 보고된 바 있는데, etomoxir와 perhexiline 등이 그 대표적인 예이다[28,59,60]. 그 밖에도 FAO의 마지막 단계에서 작용하는 효소인 3-케토아실티올분해효소(3-ketoacylthiolase)에 대한 억제제도 개발된 바 있는데 trimetazidine, ranolazine 등이 있다[61,62]. 또한, FAO 자체를 억제하기보다는 그 이후에 ETC에서 ATP가 생성되는 과정을 표적으로 하는 ETC 억제제들에 대한 항암효과도 많이 보고되었는데, 익히 당뇨병 치료약제로도 잘 알려진 metformin과 phenformin 등이 있다. Metformin은 특히나 AMPK를 활성화시키는 작용도 있어, 이를 통해 ACC가 억제되고 이어서 FAS가 차단되어 항종양 효과를 보인다는 결과도 있다[63].
Go to :
요약 및 결론
암세포는 산소가 충분한 환경에서도 해당 과정과 OXPHOS를 통한 피루브산염의 산화 과정을 분리함으로써, 탄수화물이 최대치의 ATP를 생산하는 대신 암세포 성장에 필요한 동화작용에 당대사를 특이적으로 이용한다. 이와 더불어 증식성 암대사(proliferative cancer metabolism)는 활성화된 FAS을 통해 에너지 저장, 세포막 합성, 신호전달 등을 위해 지방산을 이용한다. 그러나 때로는 진행성 암에서는 암세포의 전이와 이동을 위해 침습성 대사를 활용하게 되는데, 지방산의 대사가 중요한 역할을 한다. FAO를 통해 대사적 스트레스에 놓인 암세포에 ATP와 NADPH 공급을 할 수 있게 된다. 이처럼 암 성장의 각 단계, 혹은 암이 놓여진 환경에 맞추어 FAS와 FAO의 각 단계를 표적으로 한 치료를 고려해볼 수 있을 것이고, 다양한 암종에 적용한 여러 결과가 보고되고 있다. 두경부암은 아직 상대적으로 적용 범위가 넓지 않으나, 이미 타암종에서 적용한 긍정적인 결과들을 토대로 지방산 대사의 새로운 치료 전략 가능성을 기대해 볼 수 있을 것이다.
Go to :
 XML Download
XML Download