

 PDF
PDF Citation
Citation Print
Print



Introduction
Exposure to anesthetics induces neuronal apoptosis and long-term cognitive dysfunction in neonatal rodents [1234]. Sevoflurane is an inhaled anesthetic commonly used to induce and maintain anesthesia in pediatric patients. Recent animal studies have demonstrated that neonatal sevoflurane exposure is neurotoxic, resulting in long-term cognitive and behavioral deficits [45].
Recently, we found that the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor apocynin reduces neurotoxicity by decreasing superoxide levels and preventing mitochondrial dysfunction [6]. Furthermore, apocynin mitigates long-term memory impairment in neonatal mice exposed to sevoflurane [6]. We also found that neonatal mice exposed to sevoflurane have a reduced freezing time in the contextual fear conditioning test, probably due to decreased neuronal activation in the basolateral amygdala (BLA) [7]. A concurrent decrease in glutamatergic neurons in the BLA could explain the learning deficits caused by sevoflurane exposure [7].
Therefore, in this study, we investigated whether administration of apocynin to neonatal mice prior to sevoflurane exposure would preserve glutamatergic neurons in the BLA.
Materials and Methods
Ethical considerations and experimental mice
Ethical approval for this study was obtained from the Animal Care and Use Committee of Tokyo Medical and Dental University (approval numbers 0140163C and 0150033A). Litters of postnatal day 5 C57BL/6 male mice (average body weight 2.7 g) along with their mothers were purchased from SLC Japan, Inc., Shizuoka, Japan. The animals were housed under a 12-h light-dark cycle (lights on 08:00 to 20:00) at 21 ± 1℃. Thirty of the male pups were placed under anesthesia with sevoflurane on postnatal day 6 and then returned to their original litters. The same number of pups from each litter was used for the experiments to reduce variability related to the use of different litters. At the age of 3–4 weeks, the male pups were weaned and housed four animals to a cage. The mice had ad libitum access to water and food. Behavioral testing was performed in adulthood (11–13 weeks of age). This study used as few animals as possible. Only male mice were used to avoid potential variability caused by the estrous cycle [8].
Anesthesia protocol
On postnatal day 6, male pups (SEVO group; n = 10) were placed in a humid chamber (180 × 180 × 200 mm) with a mat heated to 38 ± 1℃, and 3% sevoflurane was delivered using a calibrated flow meter (Shimano, Tokyo, Japan) as described previously [67]. The gas was administered at 1 L/min in 40% O2 for 6 h. Pups exposed to only 40% O2 for 6 h were used as controls (NA group; n = 10). Thirty minutes prior to sevoflurane exposure, the NADPH oxidase inhibitor apocynin was administered intraperitoneally at 50 mg/kg for a total volume of 10 µl (apocynin + SEVO group; n = 10) as previously described [6]. The pups from each experimental group were returned to their litter and allowed to mature.
Fear conditioning test
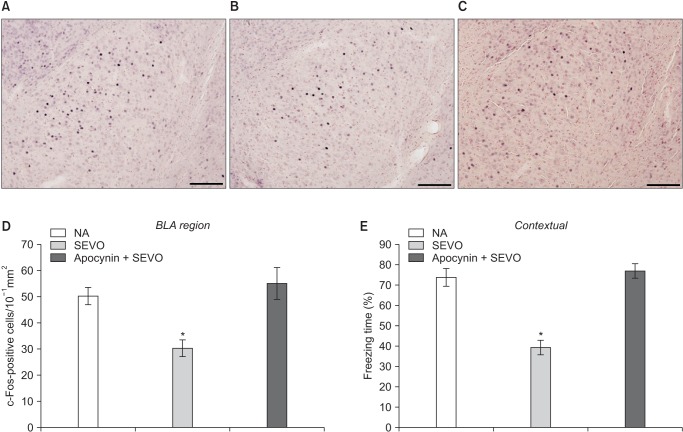
At the age of 11–13 weeks, mice were subjected to the contextual fear conditioning test. Their movement was monitored using a computerized video tracking system designed by O'Hara & Co., Ltd. (Tokyo, Japan), as described previously [67]. The test consisted of three conditioned-unconditioned stimulus pairings separated by 1-min intervals. Each pairing was as follows: unconditioned stimulus, 0.5-mA foot shock of 1-s duration; and conditioned stimulus, 60-dB white noise of 20-s duration. The unconditioned stimulus was delivered during the last seconds of the conditioned stimulus. The contextual test was performed 24 h after conditioning. The duration of freezing in response to the stimuli was automatically recorded to measure fear memory. The freezing time during total stimulation (5 min) was presented as the freezing percentage, which was compared between each group.
Tissue preparation and histopathologic evaluation
Two hours after the contextual fear conditioning test, the mice (n = 6 in each group) were deeply anesthetized with pentobarbital (50 mg/kg, intraperitoneal injection) and transcardially perfused with 4% paraformaldehyde (PFA) for 5 min. Thereafter, their brain tissue was removed and postfixed in the same buffer overnight at 4℃. Fifty-micrometer-thick floating coronal vibratome sections at 250-µm intervals (starting at bregma 2.20 mm to bregma −2.70 mm) were obtained for immunohistochemistry as previously described [7].
c-Fos staining was performed to investigate neuronal activity after the fear conditioning test. Sections were washed with phosphate-buffered saline (PBS) with 0.3% triton three times (10 min each), and endogenous peroxidase activity was blocked with 1% H2O2 (30 min) [91011]. Then, the sections were incubated with anti-c-Fos antiserum (1:1000; Santa Cruz Biotechnology, Santa Cruz, Dallas, TX, USA) diluted with 1% blocking reagent in PBS with Tween 20 (Katayama Chemistry, Osaka, Japan) overnight at 4℃. Next, the sections were incubated with peroxidase-conjugated secondary antibodies (1:1000; Abcam, Inc., Cambridge, UK) for 2 h at room temperature and washed with PBS. The c-Fos signal was visualized using a 3,3′-Diaminobenzidine (DAB)-nickel substrate (0.05% DAB, 0.05% NiSO4, 0.015% H2O2, 0.05% 1 M Tris-HCl, pH 6.7). The sections were then mounted on slides, brushed, and dried overnight. The number of c-Fos-expressing cells in the BLA was counted as described previously [7]. Briefly, four sections in the BLA from each group were carefully matched among subjects using anatomical landmarks from a brain atlas. The images were counted by a single experimenter who was blind to the treatment conditions. The number of c-Fos-expressing cells in the BLA was stated as the number of c-Fos-expressing cells/10−1 mm2 [12].
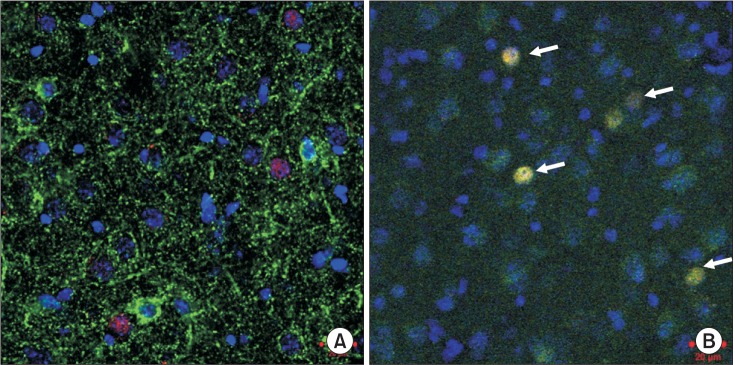
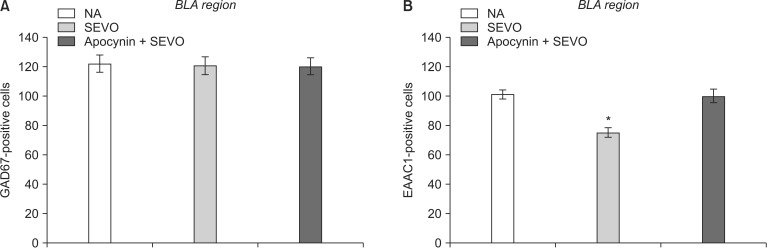
To investigate the neuron type and distribution of cells expressing c-Fos, the sections were co-incubated with anti-c-Fos antiserum and monoclonal mouse anti-Glutamic Acid Decarboxylase (GAD)67 (1:200, MAB5406; Chemicon, Temecula, CA, USA) as the GABAergic neuron marker, or with polyclonal goat anti-Excitatory Amino Acid Carrier 1 (EAAC1) (1:1000, AB1520; Chemicon) as the glutamatergic neuron marker, and then diluted in 1% blocking reagent in PBS with Tween 20 overnight at 4℃. Next, the sections were washed with PBS and co-incubated with Alexa-594-conjugated anti-rabbit IgG (1:1000; Invitrogen, Carlsbad, CA, USA) and Alexa-488 (1:1000; Invitrogen)-conjugated anti-mouse IgG or Alexa-488-conjugated anti-goat IgG (1:1000; Invitrogen) secondary antibody for 2 h at room temperature. Sections were counterstained with 4,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific Corp., Carlsbad, CA, USA) and then mounted on a slide with Fluoromount (Diagnostic BioSystems, Pleasanton, CA, USA). The positive cells in the BLA of each group were counted in a blind manner as previously described [7]. Briefly, positive cells in the BLA were counted and the four sections were fluorescently stained. The fluorescent images were examined with a microscope using the Zeiss LSM Image Browser (Carl Zeiss MicroImaging, Jena, Germany). Settings were adjusted based on the fluorescence intensity of tissue from the control group and were identical for all sections.
Statistical analysis
Statistical analysis was performed using the SPSS statistical software package (SPSS ver. 24.0; IBM, Corp., Armonk, NY, USA). The Kruskal-Wallis omnibus test was used to compare data, followed by Bonferroni post tests. Values of P < 0.05 were considered statistically significant, and data are presented as mean ± SEM.
Results
Apocynin preserved c-Fos expression in the BLA
c-Fos staining was performed to investigate neuronal activity after the fear conditioning test. c-Fos expression in the BLA was lower in the SEVO group than in the NA group (Fig. 1) [7]. Apocynin treatment preserved neuronal activity by maintaining c-Fos expression in the BLA (Fig. 1D, SEVO vs. apocynin + SEVO, P = 0.046).
Apocynin preserved glutamatergic neurons in the BLA
Recently, we showed that the cells expressing c-Fos in the BLA were glutamatergic neurons (Fig. 2) [7]. There were fewer c-Fos-expressing glutamatergic BLA neurons in the SEVO group than in the NA group, as previously described (Fig. 1) [7]. Apocynin treatment preserved neuronal activity by maintaining the glutamatergic neurons in the BLA (Fig. 3B, SEVO vs. Apocynin + SEVO, P = 0.014). Recently, we found that the NADPH oxidase inhibitor apocynin reduces neurotoxicity by decreasing the superoxide levels and preventing mitochondrial dysfunction [6]. Furthermore, apocynin improved long-term memory impairment in neonatal mice exposed to sevoflurane [6]. Together with the results of our recent studies, this study demonstrated that the administration of apocynin not only prevents the neuroapoptosis that occurs immediately after neonatal sevoflurane exposure, but also preserves the glutamatergic neurons in the BLA long after sevoflurane exposure. The number of GAD67-positive cells remained unchanged in both groups, indicating that the number of GABAergic neurons was not affected by neonatal sevoflurane exposure (Fig. 3A).
Discussion
In this study, we found that apocynin preserved c-Fos expression and glutamatergic neurons in the BLA. These results, along with our previous findings, indicate that the administration of apocynin prevents long-term memory loss in neonatal mice exposed to sevoflurane by preserving glutamatergic neurons in the BLA.
Recent studies have shown that sevoflurane exposure induces widespread apoptosis in the brain of neonates, causing severe neuronal damage in the hippocampus and amygdala, which are associated with learning and memory [59]. Specifically, after general anesthesia, glutamatergic and GABAergic neurons undergo anesthesia-induced apoptosis [10]. GABAergic neurons undergo a switch from excitatory to inhibitory roles during development [11]. In a previous study, we showed that sevoflurane exposure did not affect the number of GABAergic neurons in the BLA in adult mice and that reduced glutamatergic neurons in the BLA led to learning deficits [7].
We found that NADPH oxidase activation plays an important role in sevoflurane-induced neurotoxicity and long-term cognitive impairment [6]. Neonatal sevoflurane exposure increased NADPH oxidase subunit expression, resulting in superoxide overproduction, mitochondrial dysfunction, and apoptosis. Administration of the NADPH oxidase inhibitor apocynin prior to sevoflurane exposure prevents long-term cognitive impairment seen in adulthood. Recent animal studies have identified several therapeutic strategies that prevent the long-term learning deficits induced by neonatal sevoflurane exposure [6131415]. However, these studies mainly focused on anti-apoptotic effects, rather than the prevention of anesthesia-induced long-term learning deficits [6131415].
Recently, we found that a difference in c-Fos expression in the BLA after the fear conditioning test was associated with learning deficits induced by neonatal sevoflurane exposure and that c-Fos-expressing glutamatergic neurons within the amygdala were decreased in neonatal mice exposed to sevoflurane [7]. Expression of c-Fos reflects the neural activity after behavioral tests [161718]. Learning involves many regions of the brain [16192021]. The BLA plays a role in the acquisition and expression of learned fear [2223]. We found that a reduction in BLA glutamatergic neurons was associated with a decreased freezing time during the contextual fear conditioning test in mice exposed to sevoflurane as neonates, and that the administration of apocynin prior to sevoflurane exposure preserved the glutamatergic neurons in the BLA. We did not investigate which types of neuron tend to undergo sevoflurane-induced cell death. It may be that glutamatergic neurons are susceptible to neonatal sevoflurane exposure, and apocynin-rescued glutamatergic neurons may reduce excessive superoxide concentrations.
As our recent studies indicated, 3% sevoflurane exposure does not cause adverse cardiovascular or respiratory effects in neonatal pups [57]. The current study did not include mice administered only apocynin because apocynin alone does not induce neuroapoptosis or learning deficits in the fear conditioning test [6]. Following up on our previous studies, the current study investigated only neuronal activation in the BLA.
In conclusion, this study showed that the administration of apocynin preserved glutamatergic neurons in the basolateral amygdala of neonatal mice exposed to sevoflurane. We reported previously that apocynin prevents the decreased freezing time during the fear-conditioning test in mice exposed to sevoflurane as neonates. Therefore, apocynin may prevent neuronal apoptosis and subsequent cognitive impairment by preserving the glutamatergic neurons in the basolateral amygdala.
 XML Download
XML Download