

 PDF
PDF Citation
Citation Print
Print



Introduction
Autism is a heterogeneous neurodevelopmental disorder that affects the social communication abilities of an individual. Genetic, immunologic, and environmental factors are thought to interact during the vulnerable periods of neurogenesis [1]. Children with autism are repeatedly subjected to investigations that require the use of sedatives. These children require special consideration during their perioperative management, not only because of their clinical illness but also because of the stress on family members and the participating hospital staff [2].
Many sedatives are commonly used for pediatric procedures, especially in the population with autism. Propofol (2, 6-diisopropylphenol) is a common agent used in pediatric sedation [2]. Dexmedetomidine (Precedex) is a commonly used selective α2-adrenoreceptor agonist that offers advantages in pediatric sedation during noninvasive procedures such as magnetic resonance imaging [3].
Details of autism pathogenesis are still unclear. Gamma-aminobutyric acid type A (GABAA) [4] receptor and N-methyl-D-aspartate (NMDA) receptor [5], which is a specific type of ionotropic glutamate receptor, have been previously reported to play a role in the mechanism of autism.
However, few controlled studies [1,6], have been conducted on this cohort of patients. Recently, our research group observed a variation in propofol sedation requirements among autistic children undergoing the auditory brainstem response test when compared with others with delayed language development [7]. However, observational studies are vulnerable to confounding. Therefore, we initiated a study on an autistic rat model with the primary objective to investigate the sedation requirements by using two sedatives, propofol and dexmedetomidine, to support our observational study. The secondary objective was to explore the mechanism of autism in our model for better clarity by using GABAA and glutamate (NMDA) receptor gene expression as well as electroencephalographic (EEG) recordings in both autistic and control rats.
Materials and Methods
Study design and animal model
Animal experiments were carried out in accordance with the ethical guidelines adopted by the Faculty of Medicine, Cairo University. Fig. 1 summarizes the study design. To study the sedative requirements in autistic patients, we induced an autism-like disorder in rat pups by injecting their mothers with valproate during conception. To secure the required number of rat pups, eight adult female Sprague-Dawley rats were obtained from the animal house of the National Ophthalmology Institute and were allowed to mate. The first gestational day was recorded as the first day spermatozoa were noted in the vagina. On the 12th day of gestation, the pregnant females were randomly divided into two groups of four females each: the first group was injected subcutaneously with sodium valproate (Depakine 100 mg/ml; Sanofi-Aventis, France) at a dose of 600 mg/kg dissolved in 0.9% saline for a concentration of 100 mg/ml [8]. The other four females provided rat pups for the control group and were treated subcutaneously with saline. After delivery, the rat pups were allowed to stay with their mothers until weaning. Only male pups were used for the experiments.
Examination for autistic features and study assessments
All pups were weighed weekly until the time of the experiments to compare weight gain between the two groups (autism model and control groups). For confirming the autistic features in the autism model group, eye opening was assessed daily starting from the 7th postnatal day (PND) [9] Rats were weaned at 21 days of age; they were then transferred to the Medical Pharmacology Department, Cairo University, and were housed in groups of 2–4 in standard housing cages with free access to food and water. To prevent any possible litter effect, the rat pups from each individual mother were independently randomized into the propofol or dexmedetomidine groups. The open-field test was conducted on PND 28; each animal was placed in the center of an open-field square chamber (60 × 60 cm, with 30-cm height) [10]. Lines divided the bottom of the chamber into 16 squares. We assessed the number of grooming and rearing movements and counted the number of squares traversed by each rat during a 3-min period [11]. The video recording was used by an observer blinded to the group assignments to verify the number of squares counted.
Loss and return of righting reflex
To determine the sedative requirements, each rat was injected intraperitoneally (IP) with a single 50-mg/kg dose of propofol (Provive 1%; Claris Life sciences Limited, India) or 0.2 mg/kg dexmedetomidine hydrochloride (Precedex, 100 μg/ml; Hospira Inc., USA). The doses were guided by the recommendations of Yuan et al. [12] for propofol and Doze et al. [13] for dexmedetomidine, but the choice of doses actually used was based on preliminary pilot experiments. Time to loss of righting reflex (LORR) was measured as the time in minutes from the IP injection to the complete loss of reflex righting from the prone position. Time to return of righting reflex (RORR) was measured as the time from LORR to the time of spontaneous return to the supine position [14].
EEG recording
After return of righting from the first sedative dose, the rats were housed overnight. On the following day, 1.5 times the sedative dose was administered IP. Two minutes after LORR, electrodes were placed under the rat’s scalp: the positive electrode on one side of the head, the negative electrode on the other side, and a reference electrode at the back of the head. EEG recording was captured on Power Lab (ML866; model number 430-0820; ADInstruments, USA). Shielded, low-weight, flexible cables, connected the electrodes to the input EEG dual bioamplifier (ML408; DBS337, ADInstruments, USA). EEG was recorded on a single channel. The recorded EEG was visually inspected, and the pattern was compared between the control and autism model animals to judge the depth of sedation [15].
Hippocampal extraction
Hippocampal extraction was performed according to the method described by Spijker [16]. Decapitation was done at the end of the experiments lasted one week (five working days), and the heads were kept in the refrigerator for 10 min. Through a midline incision in the skin and bone, the parietal bones were removed and the brain was exposed, extracted, and transferred to a metal plate placed on ice. The brain was cut along its midline and the two brain halves were gently separated. The hind brain, midbrain, and the olfactory part were removed from each half, and the brain was placed such that its medial surface faced upwards. Using a spatula, the brain tissue was held and the tip of another spatula was inserted close to the corpus callosum, thalamus, and striatum. This allowed the hippocampus to be seen easily; thereafter, the spatula was put on the medial surface of the hippocampus that was separated easily from the cortex, which could be differentiated by its different color. The hippocampus was immediately placed in an Eppendorf tube and stored at (−70.0°C) until polymerase chain reaction (PCR) analysis.
Quantitative analysis of the expression of GABAA and glutamate NMDA receptor genes by using real-time PCR
Complementary DNA (cDNA) synthesis
cDNA was synthesized from 1 μg of RNA by using Super-Script III First-Strand Synthesis System (#K1621; Fermentas, USA) as described by the manufacturer. Total RNA (1 μg) was mixed with 50 μM oligo (dT) 20, 50 ng/μl random primers, and 10 mM dNTP mix in a total volume of 10 μl. The mixture was incubated at 56°C for 5 min, and then placed on ice for 3 min. The reverse transcriptase (RT) master mix containing 2 μl of 10 × RT buffer, 4 μl of 25 mM MgCl2, 2 μl of 0.1 M dithiothreitol, and 1 μl of SuperScriptⓇ III RT (200 U/μl) was added and incubated at 25°C for 10 min followed by a 50-min incubation at 50°C.
Real-time quantitative PCR
Real-time PCR amplification and analysis were performed using StepOneTM with software version 3.1 (Applied Biosystems, USA). The reaction contained SYBR Green Master Mix (Applied Biosystems). Gene-specific primer pairs (Table 1) were designed using Gene Runner (Hasting Software, Inc., USA) by utilizing RNA sequences from the GenBankⓇ is the NIH genetic sequence database gene bank. All primer sets had a calculated annealing temperature of 60°C. Real-time quantitative PCR was performed in a 25-μl reaction volume consisting of 2 × SYBR Green PCR Master Mix (Applied Biosystems), 900 nM of each primer, and 2 μl of cDNA. The amplification conditions were as follows: 2 min at 50°C, 10 min at 95°C, and 40 cycles of denaturation for 15 s and annealing/extension at 60°C for 10 min. Data from the real-time assays were analyzed using StepOneTM with software version 3.1 (Applied Biosystems, USA). Relative gene expression of the studied gene mRNAs was calculated using the comparative Cycle threshold value (Ct method). All values were normalized to those of glyceraldehyde 3-phosphate dehydrogenase, which was used as the control housekeeping gene, and reported as fold change over background levels detected in the diseased groups.
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics for Windows, Version 21.0 (IBM Corp., USA). Sample size estimation was based on an assumed mean difference in the times to LORR or RORR of 10 min and a standard deviation of 6 at a two-sided alpha of 0.05 and beta of 0.2. Numerical data were examined for normality and were presented as medians and quartiles. Groups were compared using the non-parametric Mann-Whitney test. Grooming movements/behaviour was compared as a categorical variable by using the chi-squared test. To test for the correlation of LORR and RORR with the measured GABAA and NMDA receptor gene expressions, Spearman rank correlation was used.
Results
Autistic features
Table 2 shows a comparison between the rats exposed to valproate injection and those with no exposure. No statistically significant difference was observed in weight between the two groups at birth or at the time of weaning. The rats with induced autism exhibited delayed eye opening. In the open-field test, the rats with autism traversed significantly fewer squares than did the control rats (P = 0.032). They also showed significantly fewer rearing movements than did the control rats (P = 0.016). Grooming behavior was not significantly different between the two groups.
Response to sedatives
As shown in Table 3, the rats with induced autism treated with propofol or with dexmedetomidine needed a significantly longer time period to lose their righting reflex than did the control rats. The autistic rats also needed significantly less time to RORR than did the control rats.
EEG pattern
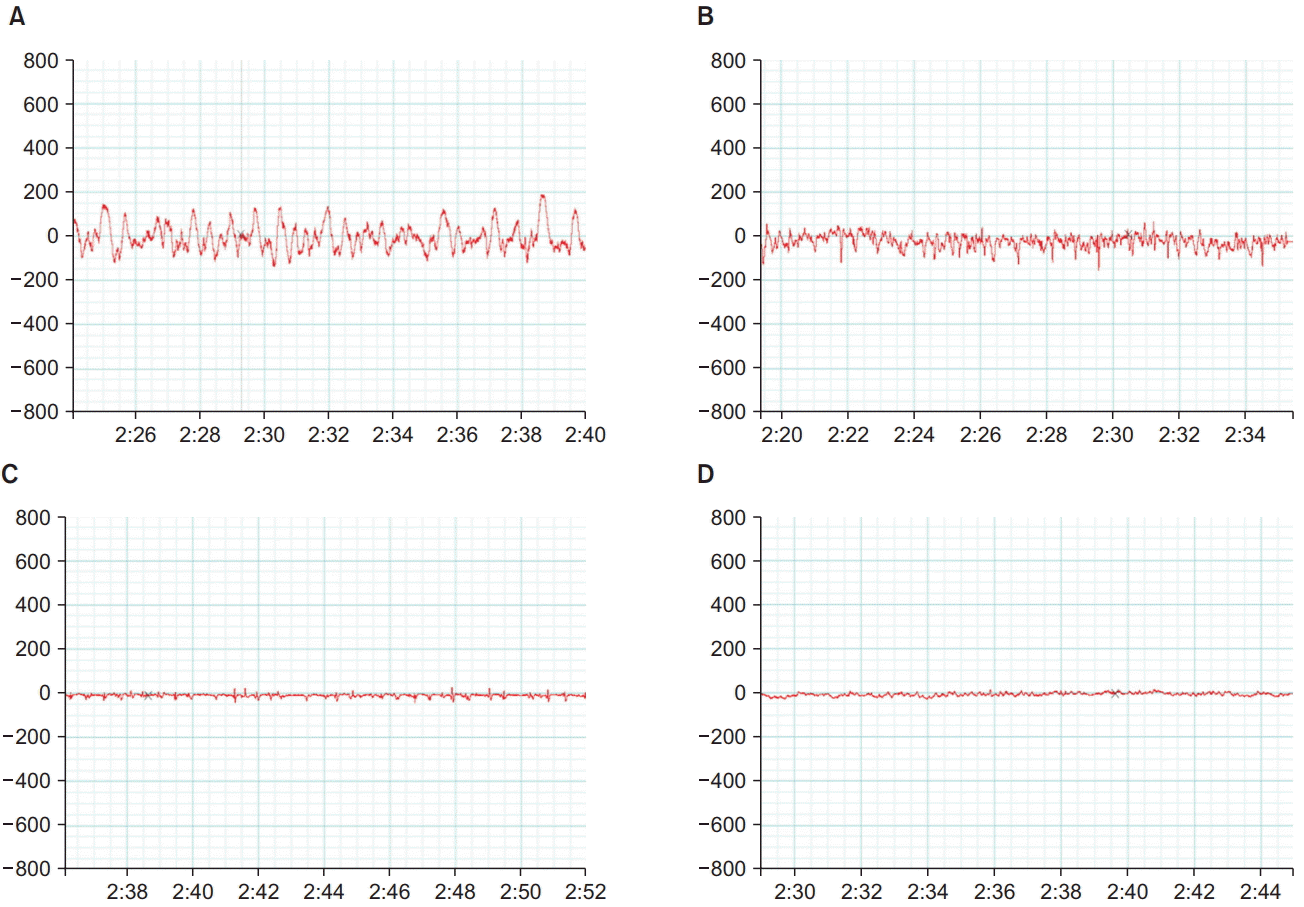
Fig. 2 shows the EEG tracings taken 2 min after LORR in response to dexmedetomidine or propofol injection in the control and autistic rats. The EEG recordings from the control rats showed the pattern of high amplitude that is usually encountered with sedation, whereas the recordings from the autistic rats showed low-amplitude, high-frequency tracings going more with an awake state.
Correlation between time to LORR and hippocampal GABAA and glutamate receptor gene expressions
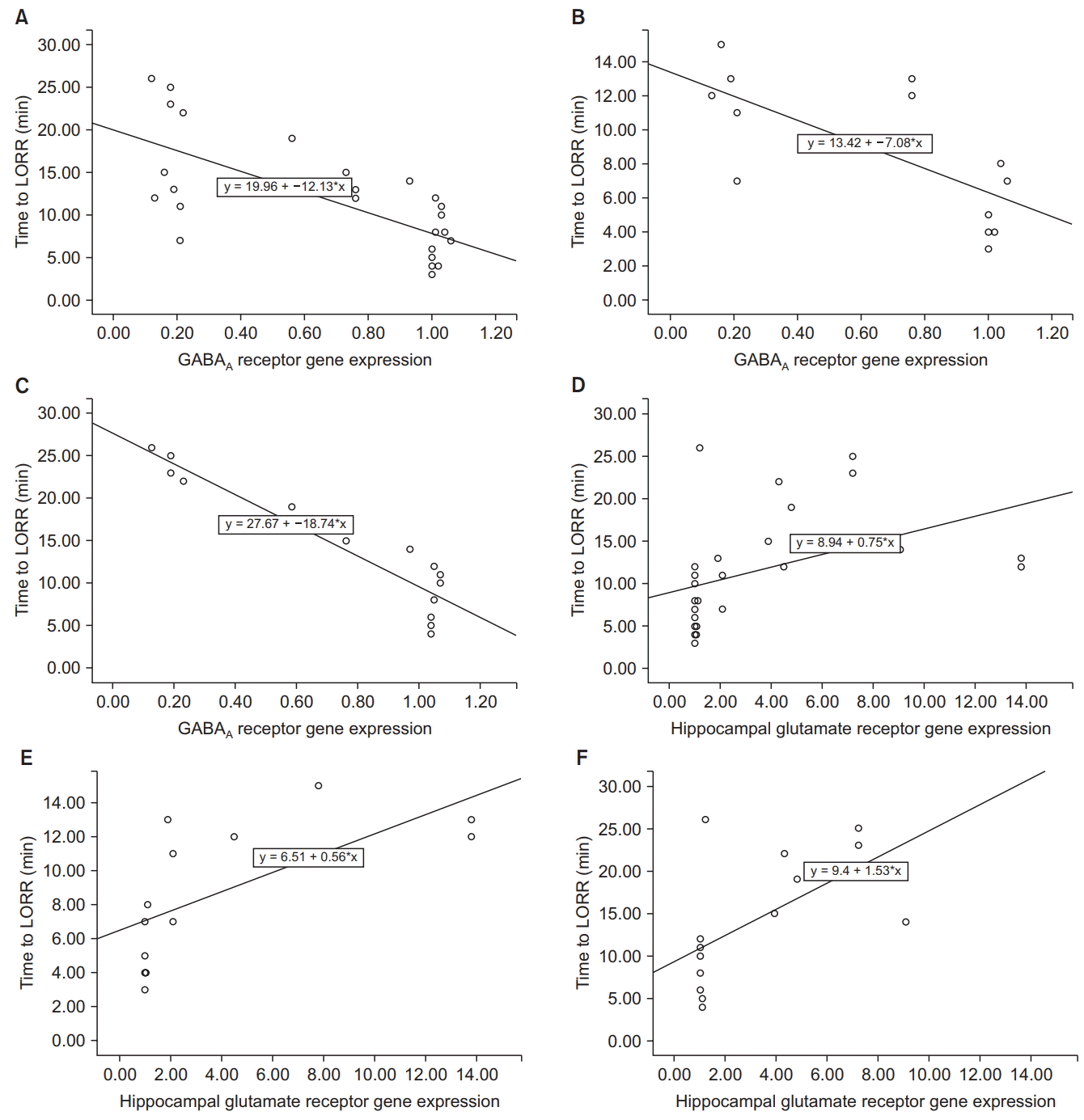
Fig. 3 shows the results of the statistical test of correlation confirming the observations on the scatter plots. A statistically significant negative correlation was observed between hippocampal GABAA receptor gene expression and the time to LORR, and a statistically significant positive correlation was observed between hippocampal glutamate receptor gene expression and the time to LORR (P < 0.05).
PCR analysis of hippocampal GABAA and glutamate receptor gene expressions
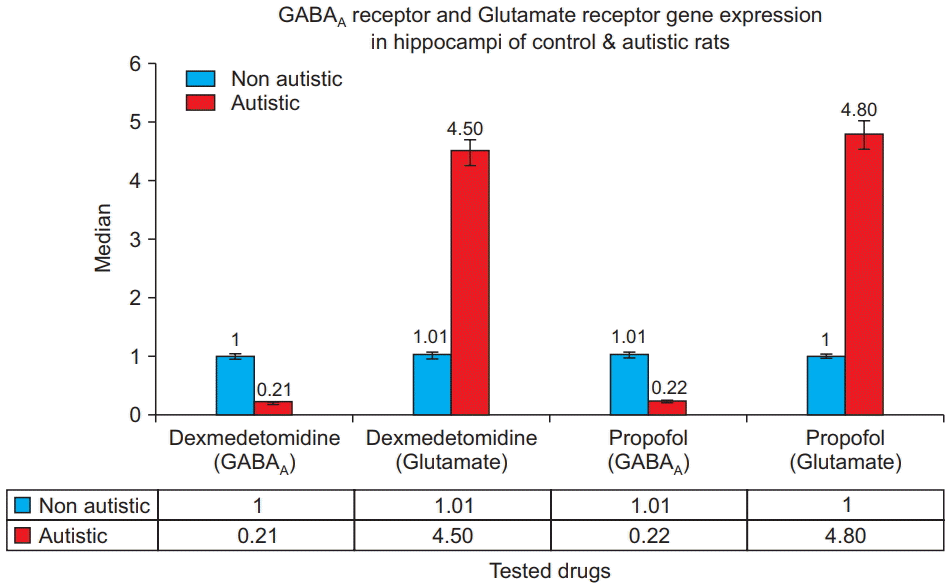
Fig. 4 shows that for both dexmedetomidine and propofol treatments, the hippocampi from the autistic rats had significantly lower GABAA receptor gene expression and significantly higher glutamate receptor gene expression than did the hippocampi from the control rats (P = 0.001). Within the control and autism model groups, no statistically significant differences were observed between GABAA gene expression in the hippocampi of rats treated with propofol and those treated with dexmedetomidine, nor was a significant difference observed in glutamate receptor gene expression (P = 0.8).
Discussion
The present study investigated the response to sedatives in an autistic rat model that has been previously used by several investigators [8,17,18]. Pregnant rats were injected with sodium valproate to induce autism in their offspring.
Clinical observational studies have suggested an association between autism and intra-uterine exposure to valproate [19]. The autistic rats showed decreased exploration of their surroundings in the open-field test and significantly less rearing movements as previously observed by Imanaka et al. [20]. This supports the validity of the model used in the current study in mimicking the autistic features reported in humans. Tas et al. [21] have reported that autism is approximately four times more common in boys than in girls. Therefore, in our autistic model, we selected male pups for investigation.
The time to LORR in rats is equivalent to that of anesthetic-induced sedation in humans [22]. Autistic rats given both sedatives (propofol and dexmedetomidine) needed a significantly longer time to achieve LORR and recovered their reflex more quickly than did the control rats. These results are consistent with those of previous clinical observations [7].
Electrical brain activity is known to change during sleep. During the awake state, EEG tracings mainly take the form of low-amplitude, high-frequency waves, and during sleep, they take the form of slow-frequency, high-amplitude waves [23]. Other researchers have also reported similar EEG patterns during sedation [24]. In the present study, we performed EEG recordings on the following day by using 1.5 times the dose of the original sedative dose to ensure sufficient depth of sedation for EEG recording. We aimed to capture the EEG changes independent of any other factors, including repeated sedative doses administered on the same day. Moreover, the control rats showed typical low-frequency, high-amplitude waves 2 min after LORR. EEG recordings of autistic rats showed a more-or-less awake pattern even after LORR. Awake patterns during sedation have been shown by MacIver and Bland in rats under isoflurane anesthesia several seconds before they recovered their righting reflex [15]. When Cusmano and Mong exposed rats prenatally to valproate and then remotely monitored their EEGs after weaning, the rats spent significantly more time awake and less time in non-rapid eye movement sleep [25]. Observations in the current study indicate that the depth of attained sedation in the autistic rats was still shallow, at 2 min after LORR. This confirms the diminished response of the autistic rats to sedatives when compared to that of normal rats.
The hippocampus is an important channel for signals transmitted to the higher brain and plays a major role in modulating behavior and long-term memory. An experimental study has linked neuronal or biochemical defects in the hippocampus to the existence of autistic features in animals [26].
In this study, we determined the level of gene expression of GABAA and glutamate receptor NMDA in the hippocampus. Animals with induced autism showed lower expression of the GABAA receptor gene and higher expression of the hippocampal glutamate receptor gene. GABAA receptors are ion channel receptors that generate fast inhibitory postsynaptic potentials that lead to the inhibition of various brain functions. Glutamate is a strong excitatory brain neurotransmitter that acts on both ionotropic (NMDA, and kainate receptors) and metabotropic receptors. Thus, changes in both GABAA and glutamate receptor gene expression point to the brain being more on the excitatory side in autistic animals. This can explain their need for a longer time to achieve an adequate depth of sedation. In fact, an imbalance of excitatory/inhibitory neurotransmission has always been a recognized feature of autism [27].
Disorders of GABAA receptors and neurons have been shown in different autism models. Sgado et al. [17] detected alterations in the gene expression and morphology of GABAminergic neurons in the cortex. Impairment of GABAminergic transmission was shown electrophysiologically by Banerjee and his colleagues [18].
In the current study, the time to LORR correlated significantly and positively with hippocampal glutamate receptor gene expression and negatively with hippocampal GABAA receptor gene expression. Alterations in the time to LORR were noted with both propofol and dexmedetomidine, even though they had distinct mechanisms of action. Propofol binds to the ligand-gated GABAA ion channel receptors and can thus be affected by the decrease in GABAA gene expression [28]. The decrease in GABAA gene expression explains the altered response to propofol. In contrast, dexmedetomidine has a very selective agonistic action on α2-adrenoceptors. By reducing norepinephrine release, dexmedetomidine decreases neuronal activity in the locus coeruleus (LC) of the brain stem and leads to sedation [29]. Two possible explanations of the delayed response of autistic rats to dexmedetomidine are postulated. The first is based on the increase in glutamate in the rat brain. Alternatively, dexmedetomidine may partially and indirectly affect GABAminergic transmission. Nelson et al. [30] suggested that the inhibition of norepinephrine release from the LC by dexmedetomidine releases the inhibitory control over the ventrolateral preoptic nucleus (VPN). The VPN releases GABA and galanin, which lead to more LC inhibition. VPN also inhibits histamine release, thereby leading to a hypnotic response.
Vlassakova and Emmanouil [1] have reported that dexmedetomidine sedation requirements have no differences between patients with autism and neurobehavioral conditions. Gimbler Berglund et al. [6] investigated the possible correlation between neurological disorders and the required propofol sedative dose for patients undergoing dental procedures as well as the recovery time. To our knowledge, the existing literature has not shown the superiority of one drug over another for the intraoperative management of children with autism. Therefore, we recommend performing future controlled studies comparing different sedatives and analyzing their safety and efficacy in children with autism.
In conclusion, the results of the present study support previous clinical observations that autism may alter the sedative requirements in children. Our biochemical analyses provide some explanation and highlight possible mechanisms for that observation. We recommend a detailed prospective clinical study.
 XML Download
XML Download