

 PDF
PDF Citation
Citation Print
Print


Introduction
Mechanical ventilation (MV), a life-saving intervention for critically ill patients with respiratory failure caused by acute lung injury (ALI), can cause an inflammatory response in both diseased and normal lungs. MV itself can also cause ventilator-induced lung injury (VILI), a form of ALI [1]. VILI results not only from barotraumas caused by alveolar disruption, capillary leakage, or lung edema, but also from biotraumas caused by the secretion of pro-inflammatory cytokines and mediators in pulmonary cells [1,2]. In particular, a complex pro- and anti-inflammatory immune reaction occurs when mechanical ventilation with high tidal volume and high inspiratory pressure is conducted on patients that require high ventilation pressure to maintain appropriate oxygenation and to remove CO2, and an imbalance eventually occurs as the inflammatory reactions progress, leading to the deterioration of ALI [3,4].
Many studies have reported that volatile anesthetics can play an important role in the protection of major organs, such as the heart and lung, through their preconditioning and postconditioning effects [5,6]. Although the mechanism through which volatile anesthetics provide organ protection is not clear, recent studies have reported that isoflurane and halothane had anti-inflammatory effects and reduced lung injury caused by lipopolysaccharide (LPS) and also reduced the pro-inflammatory cytokines secreted after MV stimulation [7,8]. Sevoflurane was also reported to have inhibited the inflammatory response after pretreatment with sevoflurane in endotoxin-induced shock and ischemia-reperfusion models [9,10]. The present study was conducted to determine whether or not sevoflurane, a commonly used volatile anesthetic, attenuates ventilator-induced lung injury in a rabbit model and also to clarify the effect sevoflurane has on the production of pro-inflammatory cytokines, such as interleukin-8 (IL-8), and the activation of intracellular signaling pathways, such as extracellular signal-regulated kinase (ERK) 1/2, p38 mitogen-activated protein kinase (MAPK), and Akt.
Materials and Methods
Animal preparation
Male New Zealand white rabbits, 1.8-2.2 kg of weight, were purchased from Samtako Science (O-San, Korea). The rabbits were kept on a 12 h light/dark cycle with access to food and water ad libitum. All experiments were conducted in accordance with institutional review board-approved protocols. The rabbits were initially anesthetized with ketamine hydrochloride (30 mg/kg, i.m.) and xylazine hydrochloride (0.2 mg/kg, i.m.). An intravenous 24 G medicut was inserted into an auricular vein to provide a route for administration of the fluid and the drugs. Normal saline was infused at a rate of 15 ml/kg/hr until the end of the study. A tracheostomy was performed aseptically, and a 3.5 mm uncuffed endotracheal tube was inserted into the trachea with spontaneous ventilation. After the start of a continuous infusion of ketamine (3 mg/kg/hr) and vecuronium bromide (0.05 mg/kg/hr) to maintain anesthesia and muscle paralysis, the lungs of the rabbit were mechanically ventilated with 50% oxygen using a pressure-controlled ventilator (PB 840, Siemen-Elema, Solna, Sweden). MV were initially set to 10 cmH2O of peak inspiratory pressure (PIP), I : E ratio of 1 : 4, and 5 cmH2O of positive end expiratory pressure (PEEP). The respiratory rate was controlled to produce an initial arterial carbon dioxide tension (PaCO2) of 35-45 mmH2O, and the rabbits were placed on a heating pad under a radiant heating lamp to keep their body temperature between 36.5 and 37.5℃ at the esophagus. The arterial catheter was placed in the aorta through the right common carotid artery cut-down to monitor arterial pressure and to harvest blood samples for blood gas analysis and assay. Then, we measure the baseline values of the mean blood pressure and arterial blood gas at 0.5 h after the start of the initial MV.
Experimental protocols
All animals were randomly assigned to one of three groups ventilated with 10 cmH2O of PIP (Sham group, n = 7); 30 cm-H2O of PIP (Control group, n = 7); or 30 cmH2O of PIP and 0.8 vol% sevoflurane (Sevorane®, Abbott, Barr, Switzerland) (Sevoflurane group, n = 7). The respiratory rate was set to maintain a normal PaCO2, respectively. These settings were maintained constant during the 5 h of MV with 0.5 fraction of inspired oxygen (FIO2). The arterial blood samples for the blood gas analysis were obtained at 0.5 and 5 h after the start of the experiment, and then all rabbits were killed at 5 h after the start of experiment by injection of an overdose of thiopental sodium. Immediately after the rabbits were killed, the thorax was opened, and the lungs were removed en bloc by observers unaware of the nature of the experiment.
Arterial blood gas analysis
Arterial blood specimens were analyzed for PaO2, PaCO2, and pH using a blood gas analyzer (GEM Premier Plus, Instrumentation Laboratory, Lexington, MA, USA).
Wet to dry weight (W/D) ratio of the lungs
The left upper lobe of the lung was weighed and then dried to a constant weight at 60℃ for over 48 h in an oven. The W/D ratio was calculated to assess tissue edema [11].
Histopathological examination
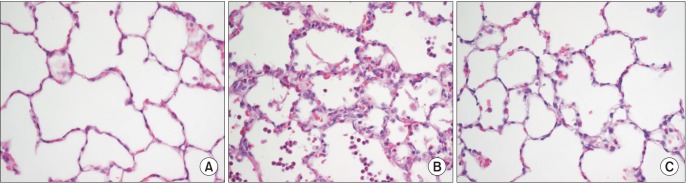
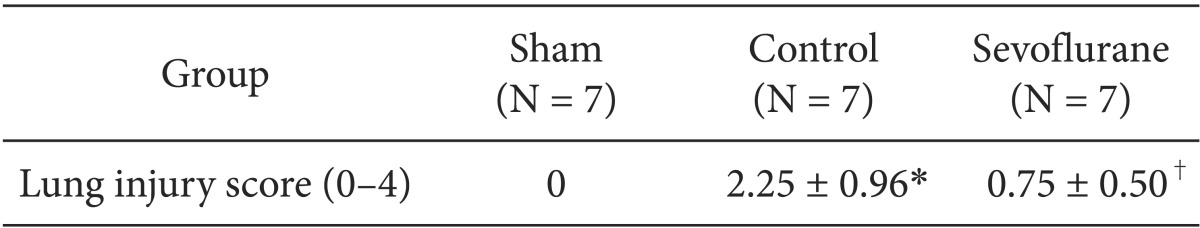
The left lower lobe was fixed by instillation of a 10% formaldehyde solution through the left lower bronchus at 20 cmH2O. The lungs were then embedded in paraffin, and the sections were stained with hematoxylin and eosin. Two observers, unaware of the nature of the experiment, scored the lung injuries under light microscopy from 0 (no damage) to 4+ (maximum damage) according to the combined assessment of alveolar congestion, hemorrhage, edema, infiltration/aggregation of neutrophils in the airspace or vessel wall, thickness of the alveolar wall, and hyaline membrane formation [11].
Preparation of bronchoalveolar lavage fluid (BALF)
BALF was harvested from the right-middle and -lower lobes. The right mainstem bronchus was the site for slow infusion and extraction of 35 ml of saline, three times. The saline contained ethylendiamine-tetraacetic acid-2Na and was cooled to 4℃ to prevent the metabolism of leukocytes. Indomethacin (5 µmol) was added to the BALF to inhibit further metabolism of arachidonic acid to prostaglandins during analysis. The BALF was centrifuged at 250 g at 4℃ for 20 min to remove the cells. The cell-free supernatant was then divided into several aliquots and was stored at -80℃ to measure the various mediators.
Measurement of IL-8
The concentration of IL-8 in BALF was quantified by using an ELISA kit obtained from R&D Systems (Minneapolis, MN, USA) according to the manufacturer's instructions.
Western blot analysis
The upper lobe of the right lung was removed from the rabbit, and whole cell extracts were prepared from lung tissue by homogenization in a protein extraction solution (PRO-PREPTM, Intron biotechnology, Seoul, Korea). Then the cell extracts were re-suspended in ice for 20 min, and debris from the lysed cells was pelleted by centrifugation at 13,000 rpm for 10 min. The protein concentration of each sample was assayed using the bicinchoninic acid (BCA) protein assay kit (Pierce, WI, USA), standardized to BSA according to the manufacturer's protocol. For Western blot analysis, 30 µg of protein were loaded and run on a 10% Tris-HCl SDS polyacrylamide gel. The protein was electrotransferred to a polyvinylidene difluoride membrane and was then blocked with 5% nonfat dry milk in 20 mM of tris buffer saline (TBS) with 0.1% Tween. After blocking, the membrane was incubated overnight at 4℃ with rabbit polyclonal specific primary Ab to p-ERK or p-p38 using a dilution of 1/1,000 in 1% BSA, followed by anti-rabbit or anti-rat IgG, Horse Radish Peroxidase (HRP)-coupled secondary antibody at a dilution of 1/2000 in 5% nonfat dry milk. After washing three times, the bands were detected using ECL Western blotting detection reagents (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and were then imaged with LAS-3000 (Life-science, Fujifilm Global, Tokyo, Japan). The membranes were then stripped using a stripping buffer (Pierce, Wisconsin, USA), and were reprobed with antibodies specific for total ERK or p38. Densitometry was performed using the Multi gauge V3.0 chemiluminescence system and analysis software (Life-science, Fujifilm Global, Tokyo, Japan) to determine the ratio between the phosphorylated and the total kinases.
Statistical analysis
The SPSS 12.0 for Windows software (SPSS Inc., Chicargo, IL, USA) was used to conduct the statistical analyses. Data from experiments are expressed as mean ± SEM. First, the importance of the significance between groups was assessed using a Kruskal-Wallis test followed by a multi comparison with Dunnett's method. A value of P < 0.05 was considered to be statistically significant.
Results
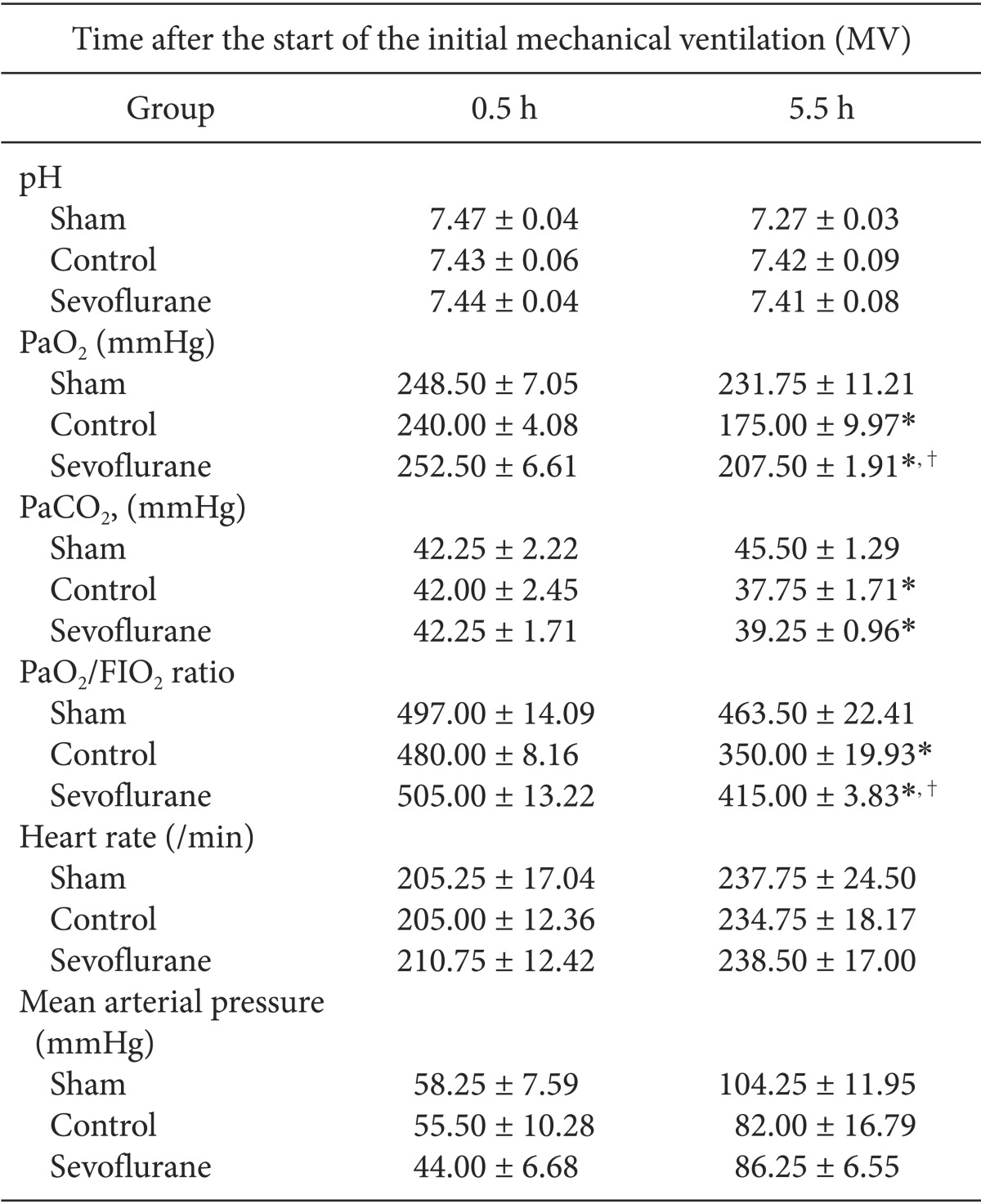
Arterial blood gas analysis
MV with high inspiratory pressure resulted in a significant decrease of PaO2/FIO2 in the control and sevoflurane groups relative to the sham group. Also, the sevoflurane group had a significantly higher PaO2/FIO2 ratio compared to that of the control group 5 h after the start of the experiment (P < 0.05, Table 1). Arterial carbon dioxide tension levels were similar in the sevoflurane and in the control group, with no significant differences (Table 1).
Circulatory variables
The heart rates and mean arterial pressure were similar between the control and the sevoflurane groups, showing no significant differences in the heart rates and in the mean arterial pressure between the study groups at any time (Table 1).
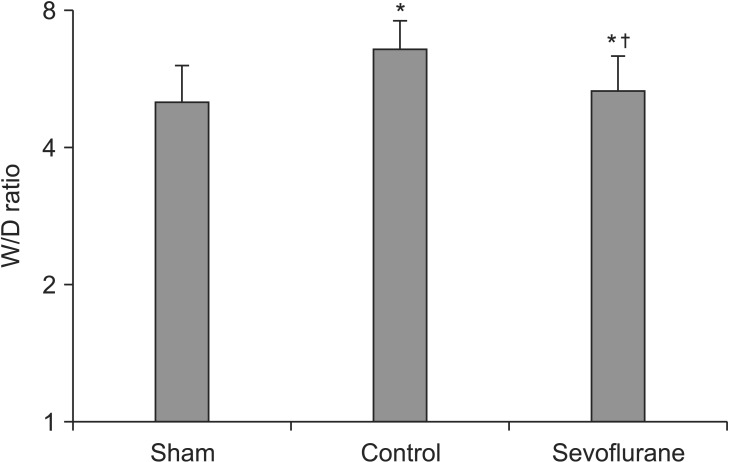
Lung wet weight to dry weight (W/D) ratio
The W/D ratio was significantly lower in the sevoflurane group than in the control group, indicating a lower lung water content [(5.36 ± 0.13) versus (6.61 ± 0.20), respectively] (P < 0.05, Fig. 2).
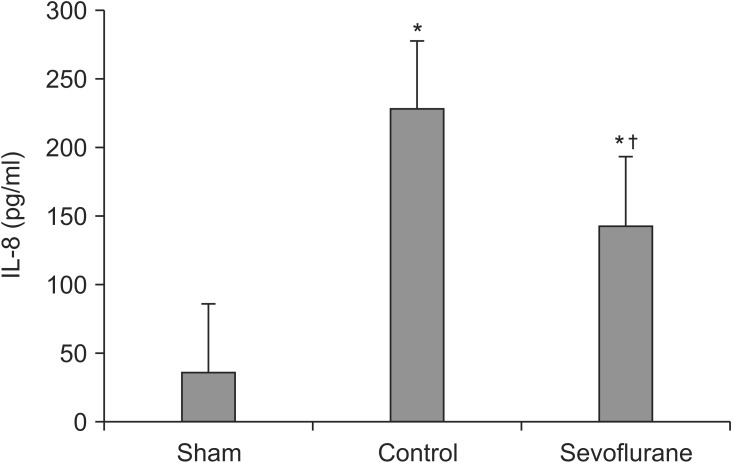
IL-8 in bronchoalveolar lavage fluid
Seventy five percent of the administered BALF was retrieved. The IL-8 levels were significantly lower in the sevoflurane group than in the control group after 5 h of MV with high inspiratory pressure [(144.08 ± 14.61) versus (228.56 ± 15.13) pg/ml, respectively] (P < 0.05, Fig. 3).
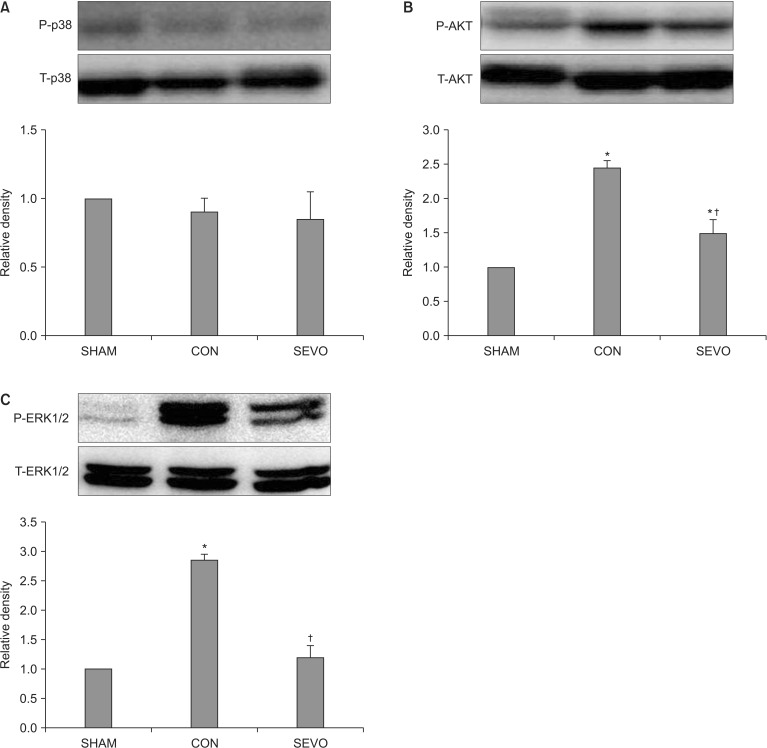
Western blot analysis
We performed Western blot analysis of the lung homogenates to determine the expression levels of several anti-apoptotic or survival protein kianses (Akt, p-Akt, p38, p-p38, ERK1/2, p-ERK1/2) in order to elucidate the molecular mechanism through which sevoflurane becomes involved in the signal transduction responsible for pulmonary protection during MV with a high inspiratory pressure. Sevoflurane significantly attenuated the expression of p-Akt and p-ERK1/2 (P < 0.05, Fig. 4). There were no significant differences in the levels of p-p38 between the control and sevoflurane groups. Western blot analyses indicated that sevoflurane-treated lungs had a decreased activation of p-Akt and p-ERK1/2, which may be associated with lung protection after MV with a high inspiratory pressure for 5 h.
Discussion
In this study, sevoflurane was observed to attenuate lung injury by decreasing the expression of the pro-inflammatory mediator IL-8 and reducing the phosphorylation of ERK1/2 and Akt in an in vivo model of VILI. These findings appear to be related to the anti-inflammatory properties of sevoflurane, including the attenuation of cytokine IL-8 production via inhibition of ERK1/2 and Akt phosphorylation in an in vivo VILI model.
Inflammatory cells and inflammatory mediators play important roles in the pathogenesis of VILI where MV with a large tidal volume or high inspiratory pressure was reported to have exhibited an inflammatory response indistinguishable from morphological and histological changes caused by the presence of bacterial lipopolysaccharides (LPS) [12]. Tsuno et al. [13] reported that lung injuries caused by over-inflation during MV resulted in alveolar hemorrhages, alveolar neutrophil infiltration, alveolar macrophage and type II pneumocyte proliferation, interstitial congestion and thickening, and interstitial lymphocyte infiltration and hyaline membrane. These findings were were similar to those in the early stage of ARDS. Inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukin (IL)-8, also play an important role in VILI [14]. However, it is not yet clear if injurious ventilation itself induces cytokine-induced pulmonary inflammation in a situation in which there is no preceding injury [15]. Whether or not TNF-α is associated with VILI exclusively as a result of MV has not been clarified in many in vivo and in vitro studies [2,9,16]. On the other hand, more consistent conclusions with respect to IL-8 have been made [16]. IL-8 release was induced, whereas TNF-α release was not seen in ex vivo studies using isolated lung cells exposed to high tidal volumes and alveolar epithelial cells exposed to cyclic pressure-stretching strains, respectively [17,18]. In addition, TNF-α release was not observed whereas IL-8 release was seen in the BALF of rats that underwent MV in vivo [16]. In this study, IL-8 significantly increased in the rabbits that had no preceding lung injuries five hours after MV with high inspiratory pressure. IL-8 increased in the group treated with sevoflurane as well, but the magnitude of the increase was less than that of the control group. Thus, IL-8 is likely to play an important role in VILI.
The cellular signaling of protein kinase B (Akt) and extracellular-signal-regulated kinases 1/2 (ERK1/2), which were reported to be associated with lung injury, was a special area of interest in this study. Increased Akt activity has been known to be associated with lung injuries and neutrophil migration to the injured site. A study on lung injury due to hemorrhaging reported that the lung was protected by attenuating inflammatory responses as a result of the inhibition of ERK phosphorylation by administering an ERK inhibitor [19]. In this molecular pathway, the up-regulation of Akt and EKR1/2 were reported to play an important role in the induction of inflammation by the ventilator and to provide the underlying mechanism for VILI [20,21]. In this in vivo study on rabbits, the phosphorylation of ERK1/2 and Akt significantly increased when the ventilator caused a lung injury, unlike in the sham group. Likewise, the phosphorylation of ERK1/2 and Akt increased more in the sevoflurane group than in the sham group, but an attenuation of the phosphorylation of ERK and Akt was more significantly seen in the sevoflurane group than in the VILI group. Taken together, putative cellular signaling pathway associated with sevoflurane - induced production of VILI is as follows : the IL-8 inhibition of sevoflurane probably occurs through pathways that converge on ERK1/2 or Akt because these kinases regulate the production of IL-8 in the inflammatory response [12].
Volatile anesthetics have attracted attention as alternatives to conventional intravenous intensive care unit (ICU) sedation since the approval of the use of the Anesthetic Conserving Devices in Europe in 2005 [22]. Relative to intravenous sedatives, however, sevoflurane-inhaled sedation has been studied in relatively small populations of patients [22,23]. Studies on sevoflurane used for ICU sedation reported that shorter extubation and ventilator times were seen when end-tidal 0.5-1.0 vol% sevoflurane was used, and no negative effects on major organ functions, such as renal function, were observed [22,23]. For example, Cho et al. [24] reported that 1.5-2.5 vol% sevoflurane attenuates systemic and pulmonary inflammatory responses induced by a cardiopulmonary bypass. Furthermore, the ideal concentration of sevoflurane for pre- or post-conditional situations has not yet been established. Lee et al. [25] reported that volatile anesthetics had anti-inflammatory properties, and the maximal effect on sevoflurane ischemic protection was achieved with 1.1 vol% in the kidney proximal tubule. A study on alveolar epithelial cells reported that a reduction of inflammatory mediators was achieved when the inflammatory mediators were stimulated by LPS for 2 h, followed by exposure to a 2.2 vol% sevoflurane concentration for 4 h [26]. In a study on the postconditioning effects of various concentrations of volatile anesthetic sevoflurane used on inflammatory mediator expression via LPS stimulation in alveolar macrophages, inflammatory mediator expression was significantly reduced with a 3.3 vol% concentration of sevoflurane but increased with a 4.4 vol% concentration of sevoflurane [6].
Several studies have contradicted this result by reporting that inflammatory mediator expression was stimulated by volatile anesthetics [27]. Kotani et al. [28] reported that the gene expression of pro-inflammatory cytokines increased and that inflammatory responses were induced within 2 h after sevoflurane inhalation during MV. Abou-Elenain [27] reported that more intrapulmonary pro-inflammatory responses were induced by sevoflurane during chest surgery. The aforementioned contradictory results are likely to have been due to the differences in each study's experimental setup, volatile anesthetics concentration, and time of exposure. In this animal study, 0.8 vol% sevoflurane was used, which was less than 1 vol%. Such a concentration is hemodynamically safe and can be clinically used for sedation.
This study had a few limitations. First, the sevoflurane concentration that would adequately inhibit inflammation was not determined. Second, the MV setting used in this study was not able to induce a significant enough lung injury to satisfy the diagnostic criteria for acute lung injury (PaO2/FIO2 ratio < 300 mmHg). Therefore, additional studies are required to resolve the aforementioned limitations.
In conclusion, these data provide evidence of the protective effect of volatile anesthetic sevoflurane on VILI. Sevoflurane prevents enhanced expression of inflammatory mediators, and the inhibition of phosphorylation of ERK1/2 and Akt is a possible pathway for protection. Although further research on the postconditioning effects is required, the inhalation of volatile anesthetic sevoflurane may provide a new and easily applicable preventive option for protection of the lungs upon acute injury.
 XML Download
XML Download